Por exemplo, água:
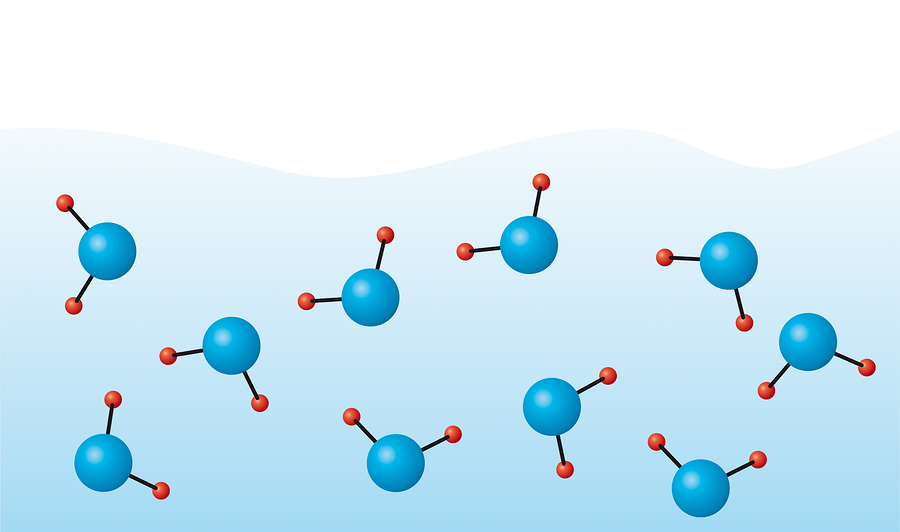
é óbvio que o hidrogênio e o oxigênio dentro de uma molécula interagem de maneira completamente diferente do que o mesmo oxigênio com o hidrogênio de uma molécula vizinha. Assim, as interações intramoleculares e intermoleculares são distinguidas. A interação intermolecular pode ser especificada por potenciais de curto alcance e pares de Coulomb, que foram discutidos em artigos anteriores. Aqui vamos nos concentrar no intramolecular.
O tipo mais comum de interação intramolecular são ligações químicas (valência). As ligações químicas são definidas pela dependência funcional da energia potencial da distância entre os átomos ligados, ou seja, de fato, pelo potencial do mesmo par. Mas, ao contrário dos potenciais de pares comuns, essa interação é especificada não para certos tipos de partículas, mas para um par específico de partículas (por seus índices). As formas funcionais mais comuns para potenciais de ligação química são potenciais harmônicos:
onde r é a distância entre as partículas, k é a constante de rigidez da ligação e r 0 é o comprimento da ligação de equilíbrio; e potencial Morse :
onde D é a profundidade do poço potencial, o parâmetro α caracteriza a largura do poço potencial.
O próximo tipo de interação intramolecular são os ângulos de ligação. Vejamos a imagem do título novamente. Por que a molécula é representada com um canto, porque as forças da eletrostática deveriam fornecer a distância máxima entre os íons de hidrogênio, que corresponde a um ângulo HOH igual a 180 °? O fato é que nem tudo está desenhado na figura. Do curso de química da escola, você pode se lembrar que o oxigênio tem mais 2 pares de elétrons solitários, a interação com a qual distorce o ângulo:

Na dinâmica molecular clássica, objetos como elétrons ou nuvens de elétrons geralmente não são introduzidos, portanto, para simular ângulos "corretos", o potencial do ângulo de ligação é usado, ou seja, dependência funcional da energia potencial nas coordenadas de 3 partículas. Um dos potenciais mais convenientes é o cosseno harmônico:
onde θ é o ângulo formado pelo tripleto de partículas, k é a constante de rigidez e θ 0 é o ângulo de equilíbrio.
Existem potenciais intramoleculares de uma ordem superior, por exemplo, ângulos de torção , mas eles são ainda mais artificiais do que os ângulos de ligação.
Adicionar interações entre partículas com índices predeterminados é trivial. Para links, armazenamos um array contendo os índices das partículas vinculadas e o tipo de interação. Damos a cada thread sua própria gama de links para processamento e pronto. Da mesma forma com ângulos de ligação. Portanto, complicaremos imediatamente a tarefa para nós mesmos: adicionaremos a capacidade de criar / remover ligações químicas e ângulos de ligação em tempo de execução. Isso imediatamente nos tira do plano da dinâmica molecular clássica e abre um novo horizonte de possibilidades. Caso contrário, você pode simplesmente baixar algo dos pacotes existentes, por exemplo LAMMPS , DL_POLY ou GROMACS , especialmente porque eles são distribuídos gratuitamente.
Agora, algum código. Vamos adicionar os campos apropriados à estrutura principal:
//bonds:
int nBond; // number of bonds
int mxBond; // maximal number of bonds
int4* bonds; // array of bonds
int* nbonds; // count of bond for a given atom
int* neighToBind; // a neighbor of a given atom for binding
int* canBind; // flags that atom[iat] can be bind
int* r2Min; // distances for the nearest neighbor (used for binding)
int* parents; // indexes of one of the atom bonded with a given
cudaBond* bondTypes;
int** def_bonds; // array[nSpec][nSpec] of default bond types
int** bindBonds; // array[nSpec][nSpec] bond types created by binding
float** bindR2; // square of binding distance [nSpec][nSpec]
//angles:
int nAngle; // number of angles
int mxAngle;
int4* angles; // array of angles
int* nangles; // number of angles for given atom
int* oldTypes;
cudaAngle* angleTypes;
int* specAngles; // [nSp] angle type formed by given species
O número de links e cantos é variável, mas quase sempre você pode estimar o máximo possível e alocar memória imediatamente ao máximo, de modo a não superalocar memória, os campos nBond e mxBond , respectivamente, significam o número atual de links e o máximo. A matriz de ligações conterá os índices dos átomos a serem ligados, o tipo de ligação e o tempo de criação da ligação (se de repente estivermos interessados em estatísticas como a vida útil média da ligação). A matriz de ângulos manterá os índices do trio de átomos que formam o ângulo de ligação e o tipo de ângulo de ligação. Os bondTypes e angleTypes matrizes conterá as características dos possíveis potenciais de ligação e ângulos. Aqui estão suas estruturas:
struct cudaBond
{
int type; // potential type
int spec1, spec2; // type of atoms that connected by this bond type
int new_type[2]; // bond type after mutation
int new_spec1[2], new_spec2[2];
int mxEx, mnEx; // flags: maximum or minimum of bond length exists
float p0, p1, p2, p3, p4; // potential parameters
float r2min, r2max; // square of minimal and maximal bond length
float (*force_eng)(float r2, float r, float &eng, cudaBond *bond); // return energy
int count; // quantity of such bonds
float rSumm; // summ of lentghs (for mean length calculation)
int rCount; // number of measured lengths (for mean length calculation)
int ltSumm, ltCount; // for calculation of lifetime
};
struct cudaAngle
{
int type; // potential type
float p0, p1, p2; // potential parameters
void (*force_eng)(int4* angle, cudaAngle* type, cudaMD* md, float& eng);
};
O campo type define a forma funcional do potencial, new_type , new_spec1 e new_spec são os índices do tipo de ligação e os tipos de átomos a serem ligados após as mudanças de ligação (quebra ou se transforma em um tipo diferente de ligação). Esses campos são representados como matrizes com dois elementos. O primeiro corresponde à situação em que o comprimento se torna menor do que r2min 1/2 , o segundo - quando ultrapassa r2max 1/2... A parte mais difícil do algoritmo é a aplicação das propriedades de todas as ligações, levando em consideração a possibilidade de sua quebra e transformação, bem como o fato de que outros fluxos poderiam quebrar ligações vizinhas, o que levou a uma mudança no tipo de átomos ligados. Deixe-me explicar usando o exemplo da mesma água. Inicialmente, a molécula é eletricamente neutra, as ligações químicas são formadas por elétrons comuns ao hidrogênio e ao oxigênio. Grosso modo, podemos dizer que as cargas nos átomos de hidrogênio e oxigênio são zero (na verdade, a densidade do elétron é deslocada para o oxigênio, portanto, há uma pequena vantagem no hidrogênio, δ +, e no oxigênio - 2δ-). Se quebrarmos a ligação, o oxigênio finalmente pegará um elétron para si e o hidrogênio o entregará. As partículas resultantes são H + e O - . No total, 5 tipos de partículas são obtidos, vamos designá-los convencionalmente: H, H + , O, O- , O 2- . Este último é formado se separarmos ambos os hidrogênios da molécula de água. Assim, as reações:
H 2 O -> H + + OH -
e
OH - -> H + + O 2- .
Especialistas em química vão me corrigir que sob condições padrão para água, o primeiro estágio de decomposição praticamente não é implementado (em equilíbrio, apenas uma molécula de 10 7dissociado em íons e, mesmo assim, não exatamente como está escrito). Mas, para a descrição de algoritmos, tais esquemas serão ilustrativos. Suponha que um fluxo processe uma ligação em uma molécula de água e outro fluxo processe a segunda ligação da mesma molécula. E aconteceu que ambos os laços precisam ser quebrados. Então, um fluxo deve transformar átomos em H + e O - , e o segundo em H + e O 2- . Mas se os fluxos o fizerem simultaneamente, no momento do início do procedimento, o oxigênio está no estado O e ambos os fluxos o convertem em O - , o que é incorreto. Precisamos prevenir tais situações de alguma forma. Diagrama de blocos de uma função que lida com uma ligação química:

Verificamos se os tipos atuais de átomos correspondem ao tipo de conexão, caso contrário, retiramos da tabela de tipos padrão (deve ser compilado antecipadamente), determinamos o quadrado da distância entre os átomos (r 2 ) e, se a conexão implica um comprimento máximo ou mínimo, verificamos se não saiu se estamos além dessas fronteiras. Se o fizermos, precisaremos alterar o tipo de conexão ou excluí-la e, em ambos os casos, alterar os tipos de átomos. Para isso, a função atomicCAS será usada- comparamos o tipo de átomo atual com o que deveria ser e neste caso o substituímos por um novo. Se o tipo do átomo já foi alterado por outro encadeamento, voltamos ao início para substituir o tipo de link. O pior cenário é se conseguirmos mudar o tipo do primeiro átomo, mas não o segundo. Já é tarde para voltar, porque depois que trocamos o primeiro átomo, outras threads já podem fazer algo com ele. Qual é a saída? Sugiro que finjamos que estamos rompendo / mudando uma conexão de um tipo diferente, e não aquela que abordamos no início. Descobrimos que tipo de conexão deveria haver entre o primeiro átomo inicial e o segundo átomo alterado e o processamos de acordo com as mesmas regras originalmente esperadas. Se neste caso o tipo de átomo mudou novamente, usaremos o mesmo esquema novamente. Está implicitamente implícito aqui,que um novo tipo de ligação tem as mesmas propriedades - quebra no mesmo comprimento, etc., e as partículas formadas durante a quebra são conforme necessário. Uma vez que esta informação é tocada pelo usuário, nós meio que transferimos a responsabilidade do nosso programa para ele, ele deve configurar tudo corretamente. O código:
__global__ void apply_bonds(int iStep, int bndPerBlock, int bndPerThread, cudaMD* md)
{
int def;
int id1, id2; // atom indexes
int old, old_spec2, spec1, spec2, new_spec1, new_spec2; // atom types
int new_bond_type;
int save_lt, need_r, loop; // flags to save lifetime, to need to calculate r^2 and to be in ‘while’ loop
int mnmx; // flag minimum or maximum
int action; // flag: 0 - do nothing, 1 - delete bond, 2 - transform bond
cudaBond *old_bnd, *cur_bnd; // old bond type, current bond type
float dx, dy, dz, r2, r;
float f, eng = 0.0f;
__shared__ float shEng;
#ifdef DEBUG_MODE
int cnt; // count of change spec2 loops
#endif
if (threadIdx.x == 0)
{
shEng = 0.0f;
}
__syncthreads();
int id0 = blockIdx.x * bndPerBlock + threadIdx.x * bndPerThread;
int N = min(id0 + bndPerThread, md->nBond);
int iBnd;
for (iBnd = id0; iBnd < N; iBnd++)
if (md->bonds[iBnd].z) // the bond is not broken
{
// atom indexes
id1 = md->bonds[iBnd].x;
id2 = md->bonds[iBnd].y;
// atom types
spec1 = md->types[id1];
spec2 = md->types[id2];
old_bnd = &(md->bondTypes[md->bonds[iBnd].z]);
cur_bnd = old_bnd;
save_lt = 0;
need_r = 1;
loop = 1;
#ifdef DEBUG_MODE
cnt = 0;
#endif
if ((cur_bnd->spec1 == spec1)&&(cur_bnd->spec2 == spec2))
{
//ok
}
else
if ((cur_bnd->spec1 == spec2) && (cur_bnd->spec2 == spec1))
{
invert_bond(id1, id2, spec1, spec2, &(md->bonds[iBnd]));
//... then ok
}
else // atom types do not correspond to bond types
{
save_lt = 1;
}
// end initial stage
while (loop)
{
if (save_lt)
{
def = md->def_bonds[spec1][spec2];
if (def == 0) // these atom types do not form a bond
{
#ifdef DEBUG_MODE
printf("probably, something goes wrong\n");
#endif
action = 1; // delete
break;
}
else
{
//! change bond type and go on
if (def < 0)
{
invert_bond(id1, id2, spec1, spec2, &(md->bonds[iBnd]));
def = -def;
}
md->bonds[iBnd].z = def;
cur_bnd = &(md->bondTypes[def]);
}
} // end if (save_lt)
// calculate distance (only once)
if (need_r)
{
dx = md->xyz[id1].x - md->xyz[id2].x;
dy = md->xyz[id1].y - md->xyz[id2].y;
dz = md->xyz[id1].z - md->xyz[id2].z;
delta_periodic(dx, dy, dz, md);
r2 = dx * dx + dy * dy + dz * dz;
need_r = 0;
}
action = 0; // 0 - just cultivate bond 1 - delete bond 2 - transform bond
if ((cur_bnd->mxEx) && (r2 > cur_bnd->r2max))
{
mnmx = 1;
if (cur_bnd->new_type[mnmx] == 0) // delete bond
action = 1;
else
action = 2; // modify bond
}
else if ((cur_bnd->mnEx) && (r2 < cur_bnd->r2min))
{
mnmx = 0;
action = 2; // at minimum only bond modification possible
}
// end select action
// try to change atom types (if needed)
if (action)
{
save_lt = 1;
new_spec1 = cur_bnd->new_spec1[mnmx];
new_spec2 = cur_bnd->new_spec2[mnmx];
//the first atom
old = atomicCAS(&(md->types[id1]), spec1, new_spec1);
if (old != spec1)
{
spec1 = old;
spec2 = md->types[id2]; // refresh type of the 2nd atom
// return to begin of the ‘while’ loop
}
else // types[id1] have been changed
{
#ifdef USE_NEWANG // save changes in atom type
atomicCAS(&(md->oldTypes[id1]), -1, spec1);
#endif
old_spec2 = spec2;
while ((old = atomicCAS(&(md->types[id2]), old_spec2, new_spec2)) != old_spec2)
{
//! the worst variant: this thread changes atom 1, other thread changes atom 2
// imagine that we had A-old bond with the same behavior
def = md->def_bonds[spec1][old];
#ifdef DEBUG_MODE
if (def == 0)
{
printf("UBEH[001]: in apply_bonds, change atom types. There are no bond types between Species[%d] and [%d]\n", spec1, old);
break;
}
#endif
if (def < 0) // spec1 -> new_spec2 spec2 -> newSpec1
{
cur_bnd = &(md->bondTypes[-def]);
new_spec2 = cur_bnd->new_spec1[mnmx];
}
else // direct order
{
cur_bnd = &(md->bondTypes[def]);
new_spec2 = cur_bnd->new_spec2[mnmx];
}
old_spec2 = old;
#ifdef DEBUG_MODE
cnt++;
if (cnt > 10)
{
printf("UBEH[002]: too many atempst to change spec2 = %d\n", spec2);
break;
}
#endif
}
#ifdef USE_NEWANG // save changes in atom type
atomicCAS(&(md->oldTypes[id2]), -1, spec2);
#endif
loop = 0;
}
//end change types
} // end if (action)
else
loop = 0; // action == 0, out of cycle
} // end while(loop)
if (action == 2)
{
new_bond_type = cur_bnd->new_type[mnmx];
if (new_bond_type < 0)
{
md->bonds[iBnd].x = id2;
md->bonds[iBnd].y = id1;
new_bond_type = -new_bond_type;
}
md->bonds[iBnd].z = new_bond_type;
cur_bnd = &(md->bondTypes[new_bond_type]);
}
// perform calculations and save mean bond length
if (action != 1) // not delete
{
r = sqrt(r2);
f = cur_bnd->force_eng(r2, r, eng, cur_bnd);
atomicAdd(&(md->frs[id1].x), f * dx);
atomicAdd(&(md->frs[id2].x), -f * dx);
atomicAdd(&(md->frs[id1].y), f * dy);
atomicAdd(&(md->frs[id2].y), -f * dy);
atomicAdd(&(md->frs[id1].z), f * dz);
atomicAdd(&(md->frs[id2].z), -f * dz);
atomicAdd(&(cur_bnd->rSumm), r);
atomicAdd(&(cur_bnd->rCount), 1);
}
else //delete bond
{
// decrease the number of bonds for atoms
atomicSub(&(md->nbonds[id1]), 1);
atomicSub(&(md->nbonds[id2]), 1);
md->bonds[iBnd].z = 0;
// change parents
exclude_parents(id1, id2, md);
}
if (save_lt)
{
keep_bndlifetime(iStep, &(md->bonds[iBnd]), old_bnd);
if (action != 1) // not delete
atomicAdd(&(cur_bnd->count), 1);
atomicSub(&(old_bnd->count), 1);
}
} // end main loop
// split energy to shared and then to global memory
atomicAdd(&shEng, eng);
__syncthreads();
if (threadIdx.x == 0)
atomicAdd(&(md->engBond), shEng);
}
No código, usei diretivas de pré-processador para permitir verificações de situações que podem surgir devido à supervisão do usuário. Você pode desligá-los para acelerar o desempenho. A função implementa o esquema acima, mas envolvida em mais um loop que percorre o intervalo de links pelos quais esse thread é responsável. Daqui em diante, o identificador do tipo de ligação pode ser negativo, isso significa que a ordem dos átomos na ligação deve ser invertida (por exemplo, a ligação OH e HO é a mesma ligação, mas no algoritmo a ordem é importante, para indicar isso eu uso índices com o oposto sinal), a função invert_bond a torna muito trivial para descrever. Função Delta_periodicaplica condições de contorno periódicas para coordenar as diferenças. Se precisarmos mudar não apenas as ligações, mas também os ângulos das ligações (diretiva USE_NEWANG ), então precisamos marcar os átomos para os quais alteramos o tipo (mais sobre isso depois). Para excluir a religação dos mesmos átomos com uma ligação, o array pais armazena o índice de um dos átomos associados aos dados (esta rede de segurança não funciona em todos os casos, mas para o meu é suficiente). Se quebrarmos algum tipo de conexão, precisaremos remover os índices atômicos correspondentes da matriz pais, isso é feito pela função exclude_parents :
__device__ void exclude_parents(int id1, int id2, cudaMD* md)
// exclude id1 and id2 from parents of each other (if they are)
// and seek other parents if able
{
// flags to clear parent
int clear_1 = 0;
int clear_2 = 0;
int i, flag;
if (md->parents[id1] == id2)
clear_1 = 1;
if (md->parents[id2] == id1)
clear_2 = 1;
i = 0;
while ((i < md->nBond) && (clear_1 || clear_2))
{
if (md->bonds[i].z != 0)
{
flag = 0;
if (clear_1)
{
if (md->bonds[i].x == id1)
{
md->parents[id1] = md->bonds[i].y;
flag = 1;
}
else if (md->bonds[i].y == id1)
{
md->parents[id1] = md->bonds[i].y;
flag = 1;
}
if (flag)
{
clear_1 = 0;
i++;
continue;
}
}
if (clear_2)
{
if (md->bonds[i].x == id2)
{
md->parents[id2] = md->bonds[i].y;
flag = 1;
}
else if (md->bonds[i].y == id2)
{
md->parents[id2] = md->bonds[i].y;
flag = 1;
}
if (flag)
{
clear_2 = 0;
i++;
continue;
}
}
}
i++;
}
// be on the safe side
if (clear_1)
md->parents[id1] = -1;
if (clear_2)
md->parents[id2] = -1;
}
A função, infelizmente, percorre todo o array de links. Aprendemos como processar e excluir links, agora precisamos aprender a criá-los. A função a seguir marca os átomos que são adequados para formar uma ligação química:
__device__ void try_to_bind(float r2, int id1, int id2, int spec1, int spec2, cudaMD *md)
{
int r2Int; // (int)r2 * const
// save parents to exclude re-linking
if (md->parents[id1] == id2)
return;
if (md->parents[id2] == id1)
return;
if (md->bindBonds[spec1][spec2] != 0)
{
if (r2 < md->bindR2[spec1][spec2])
{
r2Int = (int)(r2 * 100);
if (atomicMin(&(md->r2Min[id1]), r2Int) > r2Int) // replace was sucessfull
{
md->neighToBind[id1] = id2 + 1; // as 0 is reserved for no neighbour
md->canBind[id1] = 1;
}
// similar for the second atom
if (atomicMin(&(md->r2Min[id2]), r2Int) > r2Int) // replace was sucessfull
{
md->neighToBind[id2] = id1 + 1; // as 0 is reserved for no bind
md->canBind[id2] = 1;
}
}
}
}
A matriz bindBonds armazena informações sobre se esses tipos de átomos podem formar uma ligação e, em caso afirmativo, qual. A matriz bindR2 armazena a distância máxima entre os átomos necessária para a ligação. Se todas as condições forem favoráveis, então verificamos se os átomos de outros vizinhos são adequados para ligação, mas mais próximos.
As informações sobre a distância mais próxima ao vizinho são armazenadas no array r2Min (por conveniência, o array é do tipo int e os valores são convertidos para ele com multiplicação por uma constante, 100). Se o vizinho detectado for o mais próximo, então nos lembramos de seu índice na matriz neighToBind e definimos o sinalizador canBind... Há um perigo real de que, enquanto avançamos para a atualização do índice, outro encadeamento sobrescreve o valor mínimo, mas isso não é tão crítico. É aconselhável chamar essa função em funções que percorrem pares de átomos, por exemplo, cell_list ou all_pair , descritos na primeira parte . A própria ligação:
__global__ void create_bonds(int iStep, int atPerBlock, int atPerThread, cudaMD* md)
// connect atoms which are selected to form bonds
{
int id1, id2, nei; // neighbour index
int btype, bind; // bond type index and bond index
cudaBond* bnd;
int spec1, spec2; // species indexes
int id0 = blockIdx.x * atPerBlock + threadIdx.x * atPerThread;
int N = min(id0 + atPerThread, md->nAt);
int iat;
for (iat = id0; iat < N; iat++)
{
nei = md->neighToBind[iat];
if (nei) // neighbour exists
{
nei--; // (nei = spec_index + 1)
if (iat < nei)
{
id1 = iat;
id2 = nei;
}
else
{
id1 = nei;
id2 = iat;
}
// try to lock the first atom
if (atomicCAS(&(md->canBind[id1]), 1, 0) == 0) // the atom is already used
continue;
// try to lock the second atom
if (atomicCAS(&(md->canBind[id2]), 1, 0) == 0) // the atom is already used
{
// unlock the first one back
atomicExch(&(md->canBind[id1]), 1);
continue;
}
// create bond iat-nei
bind = atomicAdd(&(md->nBond), 1);
#ifdef DEBUG_MODE
if (bind >= md->mxBond)
{
printf("UBEH[003]: Exceed maximal number of bonds, %d\n", md->mxBond);
}
#endif
spec1 = md->types[id1];
spec2 = md->types[id2];
#ifdef USE_NEWANG // save that we have changed atom type
atomicCAS(&(md->oldTypes[id1]), -1, spec1);
atomicCAS(&(md->oldTypes[id2]), -1, spec2);
#endif
btype = md->bindBonds[spec1][spec2];
if (btype < 0)
{
// invert atoms order
md->bonds[bind].x = id2;
md->bonds[bind].y = id1;
md->bonds[bind].z = -btype;
bnd = &(md->bondTypes[-btype]);
// change atom types according the formed bond
md->types[id1] = bnd->spec2;
md->types[id2] = bnd->spec1;
}
else
{
md->bonds[bind].x = id1;
md->bonds[bind].y = id2;
md->bonds[bind].z = btype;
bnd = &(md->bondTypes[btype]);
// change atom types according the formed bond
md->types[id1] = bnd->spec1;
md->types[id2] = bnd->spec2;
}
atomicAdd((&bnd->count), 1);
md->bonds[bind].w = iStep; // keep time of the bond creation for lifetime calculation
atomicAdd(&(md->nbonds[id1]), 1);
atomicAdd(&(md->nbonds[id2]), 1);
// replace parents if none:
atomicCAS(&(md->parents[id1]), -1, id2);
atomicCAS(&(md->parents[id2]), -1, id1);
}
}
// end loop by atoms
}
A função bloqueia um átomo, depois o segundo e, se conseguir bloquear ambos, cria uma conexão entre eles. No início da função, os índices atômicos são classificados a fim de excluir a situação em que um thread bloqueia o primeiro átomo de um par e o outro bloqueia o segundo átomo do mesmo par, ambos os threads passam com sucesso na primeira verificação e falham na segunda, como resultado nenhum dos a conexão não os cria. E, finalmente, precisamos remover os links que marcamos para exclusão na função apply_bonds :
__global__ void clear_bonds(cudaMD* md)
// clear bonds with .z == 0
{
int i = 0;
int j = md->nBond - 1;
while (i < j)
{
while ((md->bonds[j].z == 0) && (j > i))
j--;
while ((md->bonds[i].z != 0) && (i < j))
i++;
if (i < j)
{
md->bonds[i] = md->bonds[j];
md->bonds[j].z = 0;
i++;
j--;
}
}
if ((i == j) && (md->bonds[i].z == 0))
md->nBond = j;
else
md->nBond = j + 1;
}
Simplesmente movemos os links "anulados" para o final do array e diminuímos o número real de links. Infelizmente, o código é serial, mas não tenho certeza se paralelizar ele trará algum efeito tangível. As funções que calculam a energia de ligação real e as forças nos átomos, que são indicadas pelos campos force_eng da estrutura cudaBond , ainda são deixadas de fora , mas são completamente análogas às funções dos potenciais de par descritos na primeira seção.
Ângulos de valência
Com ângulos de valência, apresentarei algumas suposições para tornar os algoritmos e funções mais fáceis, como resultado, eles serão ainda mais simples do que para ligações de valência. Primeiro, os parâmetros dos ângulos de ligação devem depender de todos os três átomos, mas aqui vamos assumir que o tipo de ângulo de ligação determina exclusivamente o átomo em seu vértice. Proponho o algoritmo mais simples para formar / remover cantos: sempre que mudamos o tipo de um átomo, lembramos desse fato no array oldTypes [] correspondente . O tamanho do array é igual ao número de átomos, inicialmente é preenchido com -1. Se uma função muda o tipo de um átomo, ela substitui -1 pelo índice do tipo original. Para todos os átomos que mudaram de tipo, remova todos os ângulos de ligação e passe por todas as ligações deste átomo para adicionar os ângulos correspondentes:
__global__ void refresh_angles(int iStep, int atPerBlock, int atPerThread, cudaMD *md)
// delete old angles and create new ones for atoms which change their type
{
int i, j, n, t, ang;
int nei[8]; // bonded neighbors of given atom
int cnt;
int id0 = blockIdx.x * atPerBlock + threadIdx.x * atPerThread;
int N = min(id0 + atPerThread, md->nAt);
int iat;
for (iat = id0; iat < N; iat++)
if (md->oldTypes[iat] != -1)
{
i = 0;
n = md->nangles[iat];
while (n && (i < md->nAngle))
{
if (md->angles[i].w)
if (md->angles[i].x == iat)
{
n--;
md->angles[i].w = 0;
}
i++;
}
// create new angles
t = md->specAngles[md->types[iat]]; // get type of angle, which formed by current atom type
if (t && (md->nbonds[iat] > 1)) // atom type supports angle creating and number of bonds is enough
{
// search of neighbors by bonds
i = 0; cnt = 0;
n = md->nbonds[iat];
while (n && (i < md->nBond))
{
if (md->bonds[i].z) // if bond isn't deleted
{
if (md->bonds[i].x == iat)
{
nei[cnt] = md->bonds[i].y;
cnt++;
n--;
}
else if (md->bonds[i].y == iat)
{
nei[cnt] = md->bonds[i].x;
cnt++;
n--;
}
}
i++;
}
// add new angles based on found neighbors:
for (i = 0; i < cnt-1; i++)
for (j = i + 1; j < cnt; j++)
{
ang = atomicAdd(&(md->nAngle), 1);
md->angles[ang].x = iat;
md->angles[ang].y = nei[i];
md->angles[ang].z = nei[j];
md->angles[ang].w = t;
}
n = (cnt * (cnt - 1)) / 2;
}
md->nangles[iat] = n;
// reset flag
md->oldTypes[iat] = -1;
}
}
A matriz specAngles contém os identificadores de ângulo de ligação correspondentes ao tipo de átomo fornecido. A função a seguir invoca o cálculo de energia e forças para todos os ângulos:
__global__ void apply_angles(int iStep, int angPerBlock, int angPerThread, cudaMD* md)
// apply valence angle potentials
{
cudaAngle* ang;
// energies of angle potential
float eng;
__shared__ float shEng;
if (threadIdx.x == 0)
shEng = 0.0f;
__syncthreads();
int id0 = blockIdx.x * angPerBlock + threadIdx.x * angPerThread;
int N = min(id0 + angPerThread, md->nAngle);
int i;
for (i = id0; i < N; i++)
if (md->angles[i].w)
{
ang = &(md->angleTypes[md->angles[i].w]);
ang->force_eng(&(md->angles[i]), ang, md, eng);
}
// split energy to shared and then to global memory
atomicAdd(&shEng, eng);
__syncthreads();
if (threadIdx.x == 0)
atomicAdd(&(md->engAngl), shEng);
}Bem, por exemplo, o potencial de tais ângulos, dando uma função cosseno harmônica, que pode indicar a estrutura do campo force_eng cudaAngle :
__device__ void angle_hcos(int4* angle, cudaAngle* type, cudaMD* md, float& eng)
// harmonic cosine valent angle potential:
// U = k / 2 * (cos(th)-cos(th0))^
{
float k = type->p0;
float cos0 = type->p1;
// indexes of central atom and ligands:
int c = angle->x;
int l1 = angle->y;
int l2 = angle->z;
// vector ij
float xij = md->xyz[l1].x - md->xyz[c].x;
float yij = md->xyz[l1].y - md->xyz[c].y;
float zij = md->xyz[l1].z - md->xyz[c].z;
delta_periodic(xij, yij, zij, md);
float r2ij = xij * xij + yij * yij + zij * zij;
float rij = sqrt(r2ij);
// vector ik
float xik = md->xyz[l2].x - md->xyz[c].x;
float yik = md->xyz[l2].y - md->xyz[c].y;
float zik = md->xyz[l2].z - md->xyz[c].z;
delta_periodic(xik, yik, zik, md);
float r2ik = xik * xik + yik * yik + zik * zik;
float rik = sqrt(r2ik);
float cos_th = (xij * xik + yij * yik + zij * zik) / rij / rik;
float dCos = cos_th - cos0; // delta cosinus
float c1 = -k * dCos;
float c2 = 1.0 / rij / rik;
atomicAdd(&(md->frs[c].x), -c1 * (xik * c2 + xij * c2 - cos_th * (xij / r2ij + xik / r2ik)));
atomicAdd(&(md->frs[c].y), -c1 * (yik * c2 + yij * c2 - cos_th * (yij / r2ij + yik / r2ik)));
atomicAdd(&(md->frs[c].z), -c1 * (zik * c2 + zij * c2 - cos_th * (zij / r2ij + zik / r2ik)));
atomicAdd(&(md->frs[l1].x), c1 * (xik * c2 - cos_th * xij / r2ij));
atomicAdd(&(md->frs[l1].y), c1 * (yik * c2 - cos_th * yij / r2ij));
atomicAdd(&(md->frs[l1].z), c1 * (zik * c2 - cos_th * zij / r2ij));
atomicAdd(&(md->frs[l2].x), c1 * (xij * c2 - cos_th * xik / r2ik));
atomicAdd(&(md->frs[l2].y), c1 * (yij * c2 - cos_th * yik / r2ik));
atomicAdd(&(md->frs[l2].z), c1 * (zij * c2 - cos_th * zik / r2ik));
eng += 0.5 * k * dCos * dCos;
}
Não darei uma função para remover cantos "anulados", não difere fundamentalmente de clear_bonds .
Exemplos de
Sem fingir ser preciso, tentei representar a montagem de moléculas de água a partir de íons individuais. Os potenciais emparelhados foram definidos arbitrariamente na forma do potencial de Buckingham e, em seguida, adicionou a capacidade de criar ligações na forma de um potencial harmônico, com uma distância de equilíbrio igual ao comprimento da ligação HO na água, 0,96 Å. Além disso, quando o segundo próton foi ligado ao oxigênio, um ângulo de ligação com o ápice no oxigênio foi adicionado. Após 100.000 passos, as primeiras moléculas surgiram de íons espalhados aleatoriamente. A figura mostra as configurações inicial (esquerda) e final (direita):

Você pode configurar um experimento como este: deixe a princípio todos os átomos serem iguais, mas quando eles estão próximos uns dos outros, eles formam uma ligação. Deixe os átomos ligados formarem outra ligação com um átomo livre ou com outra molécula ligada semelhante. Como resultado, obtemos algum tipo de auto-organização, onde os átomos se alinham em cadeias:

Comentários finais
- Aqui usamos apenas um critério de ligação - distância, embora em princípio possa haver outros, por exemplo, a energia do sistema. Na realidade, quando uma ligação química é formada, via de regra, a energia é liberada na forma de calor. Isso não é levado em consideração aqui, mas você pode tentar implementá-lo, por exemplo, alterar a velocidade da partícula.
- As interações entre as partículas por meio do potencial de ligação química não negam o fato de que as partículas ainda podem interagir por meio dos potenciais de pares intermoleculares e da interação de Coulomb. Seria possível, é claro, não calcular as interações intermoleculares para átomos ligados, mas isso, no caso geral, requer longas verificações. Portanto, é mais fácil definir o potencial de ligação química de forma que sua soma com outros potenciais dê a função desejada.
- A implementação paralela da ligação de partículas não só dá um ganho de velocidade, mas também parece mais realista, uma vez que as partículas competem entre si.
Bem, existem vários projetos em Habré que são muito próximos a este: