
A matemática é freqüentemente chamada de "linguagem da ciência". É adequado para o processamento quantitativo de quase todas as informações científicas, independentemente do seu conteúdo. E com a ajuda do formalismo matemático, cientistas de diferentes áreas podem, até certo ponto, "entender" uns aos outros. Hoje, uma situação semelhante está surgindo com a Ciência da Computação. Mas se a matemática é a linguagem da ciência, então o CS é o seu canivete suíço. Na verdade, é difícil imaginar a pesquisa moderna sem analisar e processar grandes quantidades de dados, cálculos complexos, modelagem de computador, visualização e o uso de software e algoritmos especiais. Vejamos algumas "histórias" interessantes quando diferentes disciplinas usam métodos de CS para resolver seus problemas.
Bioinformática: das placas de Petri à biologia In silico
A bioinformática pode ser considerada um dos exemplos mais marcantes da interseção da ciência da computação e outras disciplinas. Esta ciência lida com a análise de dados biológicos moleculares usando métodos de computador. A bioinformática como uma direção científica separada apareceu no início dos anos 70 do século passado, quando as sequências de nucleotídeos de pequenos RNAs foram publicadas pela primeira vez e os algoritmos para prever sua estrutura secundária (o arranjo espacial dos átomos em uma molécula) foram criados.
Uma nova era da bioinformática começou com o Projeto Genoma Humano, que visa determinar a sequência de nucleotídeos no DNA humano e identificar genes no genoma. O custo do sequenciamento de DNA (sequenciamento de nucleotídeos) caiu várias ordens de magnitude. Isso levou a um grande aumento no número de sequências em bancos de dados públicos. O gráfico a seguir mostra o crescimento do número de sequências no banco de dados público do GenBank de dezembro de 1982 a fevereiro de 2017 em escala semilogista. Para que os dados acumulados se tornem úteis, eles precisam ser analisados de alguma forma.

Crescimento do número de sequências no GenBank de dezembro de 1982 a fevereiro de 2017. Fonte: www.ncbi.nlm.nih.gov/genbank/statistics
Um dos métodos de análise de sequência em bioinformática é o alinhamento de sequência. A essência do método reside no fato de que as sequências de monômeros de DNA, RNA ou proteínas são colocadas umas sobre as outras de forma a ver áreas semelhantes. A semelhança nas estruturas primárias (ou seja, sequências) de duas moléculas pode refletir sua relação funcional, estrutural ou evolutiva. Uma vez que uma sequência pode ser representada como uma string com um alfabeto específico (4 nucleotídeos para DNA e 20 aminoácidos para proteína), o alinhamento acaba sendo uma tarefa combinatória de CS (por exemplo, o alinhamento de linha também é usado no processamento de linguagem natural - PNL). No entanto, o contexto da biologia adiciona alguma especificidade ao problema.
Vejamos o alinhamento usando proteínas como exemplo. Um resíduo de aminoácido na proteína corresponde a uma letra do alfabeto latino na sequência. As strings são escritas uma abaixo da outra para obter a melhor correspondência. Os elementos correspondentes estão um abaixo do outro, as "lacunas" são substituídas por "-" (lacuna). Eles designam indel , isto é, o local de possível inserção (introdução em uma molécula de um ou mais nucleotídeos ou aminoácidos) e deleções ("eliminação" de um nucleotídeo ou aminoácido).

Um exemplo do alinhamento das sequências de aminoácidos de duas proteínas. Leucina (L) e isoleucina (I), que são isômeros, estão destacados em azul - tal substituição na maioria dos casos não afeta a estrutura da proteína
No entanto, como você pode determinar se o alinhamento é ideal? A primeira coisa que vem à mente é estimar o número de correspondências: quanto mais correspondências, melhor. No entanto, no contexto da biologia, isso não é totalmente verdade. As substituições (substituições de um aminoácido por outro) são desiguais: algumas substituições (por exemplo, S e T, D e E são resíduos que diferem na estrutura por exatamente um átomo de carbono) praticamente não afetam a estrutura das proteínas. Mas a substituição da serina pelo triptofano mudará muito a estrutura da molécula. Um critério quantitativo (peso ou pontuação) é inserido para determinar se a compensação é a melhor possível. Para avaliar as substituições, são utilizadas as chamadas matrizes de substituição, com base nas estatísticas de substituição de aminoácidos em proteínas com uma estrutura conhecida. Quanto maior for o número na intersecção das letras correspondentes, maior será a pontuação.

Novas matrizes de substituição aparecem periodicamente. Aqui está a matriz BLOSUM62.
A pontuação também leva em consideração a presença de deleções. Normalmente, a penalidade para “abrir” uma exclusão é várias ordens de magnitude maior do que para “continuar”. Isso se deve ao fato de que uma seção de várias lacunas consecutivas é considerada uma mutação, e várias lacunas em diferentes lugares são consideradas várias. No exemplo abaixo, o primeiro par de sequências é mais semelhante do que o segundo, porque no primeiro caso, as sequências são formalmente separadas por um evento evolutivo:

Agora, sobre os próprios algoritmos de alinhamento. Existem dois tipos de alinhamento emparelhado (localizando áreas semelhantes de duas sequências): global e local. O alinhamento global implica que as sequências são homólogas (semelhantes) ao longo de todo o seu comprimento. Inclui ambas as sequências em sua totalidade. No entanto, com essa abordagem, áreas semelhantes nem sempre são bem definidas se houver poucas delas. O alinhamento local é usado se as sequências forem mantidas como homólogas (por exemplo, devido à recombinação) e sites não relacionados. Mas nem sempre pode entrar na área de interesse, além disso, existe a possibilidade de encontrar uma área semelhante acidental. Para obter um alinhamento par a par, métodos de programação dinâmica são usados (resolver um problema dividindo-o em várias subtarefas idênticas conectadas recorrentemente). Em programas para alinhamento global, o algoritmo Needleman-Wunsch é freqüentemente usado e, para alinhamento local, o algoritmo Smith-Waterman . Você pode ler mais sobre eles seguindo os links.
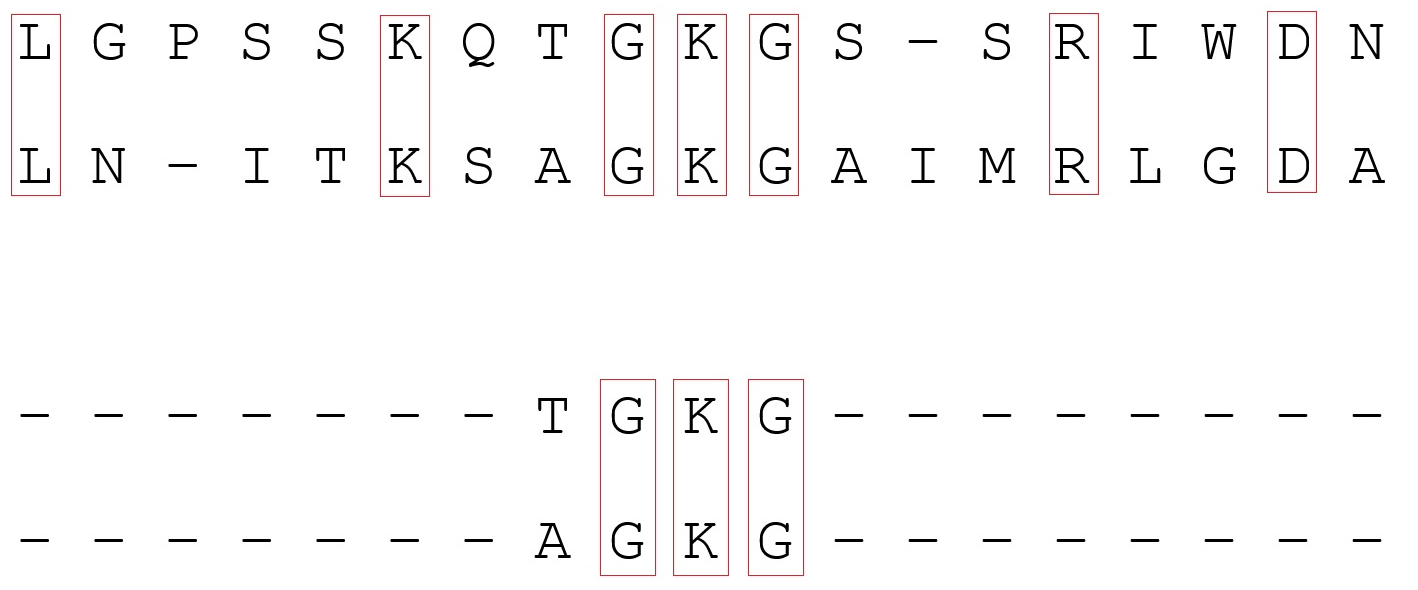
Exemplo de alinhamento: a parte superior é global, a parte inferior é local. No primeiro caso, o alinhamento ocorre ao longo de todo o comprimento das sequências, no segundo, algumas regiões homólogas são encontradas.
Como você pode ver, a tarefa biológica pode ser bastante reduzida à tarefa de CS. O alinhamento de pares usando os algoritmos mencionados requer cerca de m * n de memória adicional (m, n são os comprimentos das sequências), que os computadores domésticos modernos podem controlar facilmente. No entanto, a bioinformática também tem tarefas mais não triviais, por exemplo, alinhamento múltiplo (alinhamento de várias sequências) para a reconstrução de árvores filogenéticas.... Mesmo se compararmos 10 proteínas muito pequenas com um comprimento de sequência de cerca de 100 caracteres, uma quantidade inaceitavelmente grande de memória adicional será necessária (a dimensão da matriz é 100 ^ 10). Portanto, neste caso, o alinhamento é baseado em várias heurísticas.
Modelando a estrutura em grande escala do universo
Ao contrário da biologia, a física está lado a lado com a ciência da computação desde os primeiros dias dos computadores. Antes da criação dos primeiros computadores, a palavra "computador" (calculadora) era chamada de posição especial - eram pessoas que realizavam cálculos matemáticos em calculadoras. Assim, durante o Projeto Manhattan, o físico Richard Feynman era o gerente de toda uma equipe de "calculadoras" que processava equações diferenciais em máquinas de somar.

"Sala de informática" do Flight Research Center. Armstrong. EUA, 1949
No momento, os métodos de CS são amplamente usados em vários campos da física. Por exemplo, a física computacional estuda algoritmos numéricos para resolver problemas físicos para os quais uma teoria quantitativa já foi desenvolvida. Em situações em que a observação direta de objetos é difícil (isso geralmente acontece na astronomia), a modelagem por computador vem em auxílio dos cientistas. Exatamente esse caso é o estudo da estrutura em larga escala do Universo : as observações de objetos distantes são difíceis devido à absorção da radiação eletromagnética no plano da Via Láctea, de modo que a modelagem tornou-se o principal método de pesquisa.

,
Uma das tarefas da cosmologia moderna é explicar a imagem observada da diversidade de galáxias e sua evolução. No nível qualitativo, os processos físicos que ocorrem nas galáxias já são conhecidos, portanto, os esforços dos cientistas estão voltados para a obtenção de previsões quantitativas. Isso permitirá responder a uma série de questões fundamentais, por exemplo, sobre as propriedades da matéria escura. Mas, antes de isolar as manifestações observadas da matéria escura, é necessário entender o comportamento da matéria comum. Em uma escala enorme (vários milhões de anos-luz), a matéria comum se comporta efetivamente da mesma maneira que a escuridão: está sujeita a uma força de gravidade, você pode esquecer a pressão do gás. Isso torna relativamente fácil simular a evolução da estrutura em grande escala do Universo (métodos numéricos,contendo apenas matéria escura ou semelhante a poeira e reproduzindo bem a estrutura em grande escala da distribuição das galáxias, começou a se desenvolver a partir da década de 1980).
A matéria escura é modelada da seguinte maneira. O cubo virtual, com centenas de milhões de anos-luz de tamanho, está quase uniformemente preenchido com partículas de teste - corpos. Desde o início, pequenas inomogeneidades estiveram presentes no Universo, das quais surgiu toda a estrutura observada, portanto, o preenchimento é "quase uniforme". Então as partículas começam a “viver suas próprias vidas” sob a influência da gravidade: o problema de N corpos está resolvido . As partículas que escaparam do cubo são transferidas para a face oposta, e as forças gravitacionais também se propagam com a transferência. Graças a isso, o cubo se torna, por assim dizer, infinito, como o universo.

Trajetórias aproximadas de três corpos idênticos localizados nos vértices de um triângulo não isósceles e com velocidades iniciais nulas
Um dos modelos numéricos mais famosos desse tipo é o Millenium , que tem um tamanho de cubo de mais de 1,5 bilhão de anos-luz e cerca de 10 bilhões de partículas. Nos anos seguintes, vários modelos maiores foram feitos: o Horizon Run com o lado do cubo 4 vezes maior que o Millenium, e o Dark Sky com 16 vezes o Millenium. Esses e outros modelos semelhantes têm desempenhado um papel fundamental em projetos para validar o modelo Lambda-CDM agora geralmente aceito. (Um universo contendo cerca de 70% de energia escura, 25% de matéria escura e 5% de matéria comum).

Millenium: , ; — . .
A redução da escala causa problemas na combinação de observações e modelos numéricos com uma matéria escura. Em uma escala menor (a escala de propagação das ondas de choque das supernovas), a matéria não pode mais ser considerada empoeirada. É preciso levar em consideração a hidrodinâmica, o resfriamento e o aquecimento do gás por radiação e muito mais. Para levar em consideração todas as leis da física na modelagem, algumas simplificações são feitas: por exemplo, você pode quebrar o cubo do modelo em uma rede de células (física de sub-rede) e assumir que, quando uma certa densidade e temperatura na célula é atingida , parte do gás se transformará instantaneamente em uma estrela. Esta classe de modelos inclui os projetos EAGLE e illustris . Um dos resultados desses projetos é a reprodução da relação Tully-Fisher. entre a luminosidade da galáxia e a velocidade de rotação do disco.
Lingüística e aprendizado de máquina: um passo mais perto de resolver um mistério de 4.000 anos
Os métodos de CS encontram aplicações em áreas mais inesperadas, por exemplo, no estudo de línguas antigas e sistemas de escrita. Assim, um estudo realizado por um grupo de cientistas liderado por Rajesh P.N. Rao, professor da Universidade de Washington, lançou luz sobre o mistério da escrita do Vale do Indo.
A escrita do Indo, usada entre 2600-1900 aC no que hoje é o Paquistão Oriental e o noroeste da Índia, pertencia a uma civilização não menos complexa e misteriosa do que seus contemporâneos mesopotâmicos e egípcios. Restam muito poucas fontes escritas dele: os arqueólogos encontraram apenas cerca de 1.500 inscrições únicas em fragmentos de cerâmica, tabuinhas e selos. As letras mais longas têm apenas 27 caracteres.

Inscrições em focas do Vale do Indo
Na comunidade científica, havia várias hipóteses sobre os "símbolos misteriosos". Alguns especialistas consideram os símbolos nada mais do que apenas "belas imagens". Assim, em 2004, o lingüista Steve Farmer publicou um artigo no qual argumentava que a escrita do Indo nada mais era do que símbolos políticos e religiosos. Sua versão, embora polêmica, ainda encontrou adeptos.
Rajesha P.N. Rao, um cientista de aprendizado de máquina, leu sobre a escrita do Indo no ensino médio. Um grupo de cientistas sob sua liderança decidiu conduzir uma análise estatística de documentos confiáveis existentes. No decorrer da pesquisa usando cadeias de Markov(uma das primeiras disciplinas em que as cadeias de Markov encontraram aplicação prática foi a crítica textual) a entropia condicional foi comparada símbolos da escrita do Indo com a entropia de sequências de sinais linguísticas e não linguísticas. Entropia condicional é a entropia de um alfabeto para o qual as probabilidades de uma letra após a outra são conhecidas. Vários sistemas foram selecionados para comparação. Os sistemas linguísticos incluíam: escrita logográfica suméria, antigo Tamil Abugida, sânscrito do Rig Veda, inglês moderno (palavras e letras foram estudadas separadamente) e a linguagem de programação Fortran. Os sistemas não linguísticos foram divididos em dois grupos. O primeiro incluiu sistemas com uma ordem rígida de sinais (conjunto artificial de sinais nº 1), o segundo - sistemas com uma ordem flexível (proteínas de bactérias, DNA humano, conjunto artificial de sinais nº 2). Como resultado, descobriu-se que a escrita proto-indiana acabou sendo moderadamente ordenada, como a escrita das línguas faladas:a entropia dos documentos existentes é semelhante à entropia das escritas sumérias e tâmil.

Entropia condicional para vários sistemas linguísticos e não linguísticos
Este resultado refutou a hipótese sobre o uso ornamental de signos. E embora os métodos de CS tenham ajudado a confirmar a versão de que os símbolos do Vale do Indo são provavelmente um sistema de escrita, o assunto ainda não foi decifrado.
Conclusão
Claro, muitas áreas onde os métodos de CS encontram aplicação estão fora dos limites. É simplesmente impossível revelar em um artigo como a ciência moderna depende da tecnologia de computador. No entanto, espero que os exemplos fornecidos mostrem como diferentes problemas podem ser resolvidos, incluindo por métodos de CS.
Os servidores em nuvem da Macleod são rápidos e seguros.
Cadastre-se pelo link acima ou clicando no banner e ganhe 10% de desconto no primeiro mês de aluguel de um servidor de qualquer configuração!
