
Em um artigo publicado recentemente na Physical Review Research, demonstramos como o aprendizado profundo simplifica a solução de equações mecânicas quânticas fundamentais para sistemas reais. Ao mesmo tempo, não apenas uma questão científica fundamental é resolvida, mas também se abrem perspectivas para a utilização prática dos resultados obtidos no futuro.
Os pesquisadores serão capazes de criar protótipos de novos materiais e compostos in silico antes de tentar sintetizá-los em laboratório. Também postou o códigodeste estudo; assim, as equipes de física e química computacional podem desenvolver seu trabalho e aplicá-lo a uma variedade de problemas. Como parte do estudo, uma nova arquitetura de rede neural, Fermionic Neural Network ou FermiNet, foi desenvolvida, que é adequada para simular o estado quântico de grandes coleções de elétrons - e todas as ligações químicas são baseadas em elétrons. FermiNet demonstrou pela primeira vez como usar o aprendizado profundo para calcular a energia de átomos e moléculas do zero. O modelo resultante revelou-se preciso o suficiente para aplicação prática e, na época da publicação do artigo original (outubro de 2020), continuava sendo o método de rede neural mais preciso usado na indústria. É assumidoque os métodos e ferramentas associados podem ser úteis na solução de problemas fundamentais nas ciências naturais. Os autores da FermiNet já estão usando em seus trabalhos sobre a convolução de proteínas , a dinâmica de compostos vítreos , a cromodinâmica quântica em uma rede e em muitos outros projetos ajudando a traduzir esses desenvolvimentos em prática.
Uma breve história da mecânica quântica
Ao mencionar "mecânica quântica", é provável que você confunda o interlocutor com este tópico como nenhum outro. Imediatamente lembro-me de imagens como o gato de Schrödinger, que paradoxalmente pode estar vivo e morto ao mesmo tempo, assim como partículas elementares que são corpúsculos e ondas. Em um sistema quântico, uma partícula como um elétron não tem uma localização específica, ao contrário da situação na física clássica. Na física quântica, a posição de um elétron é descrita por uma nuvem de probabilidades - isto é, é espalhada por todos aqueles pontos, em cada um dos quais um elétron pode aparecer. Por causa desse absurdo estado de coisas, Richard Feynman achou possível afirmar: "Acho que posso dizer com segurança que ninguém entende a mecânica quântica."
Apesar de toda essa estranheza assustadora, a essência da teoria pode ser expressa em apenas algumas equações simples. A mais famosa delas, a equação de Schrödinger, descreve o comportamento de partículas em uma escala quântica da mesma forma que as equações de Newton descrevem o comportamento de corpos em escalas macroscópicas mais familiares. Embora a interpretação desta equação obrigue qualquer pessoa a agarrar a cabeça, seu componente matemático é muito mais fácil de usar, devido ao qual nasceu o famoso "cale a boca e conte" do famoso professor, com o qual eles lutaram contra as incômodas questões filosóficas dos alunos.
Essas equações são suficientes para descrever o comportamento de toda matéria familiar a nós no nível dos átomos e núcleos. Um componente ilógico da mecânica quântica está por trás de todos os tipos de fenômenos exóticos: supercondutividade, superfluidez, laser e semicondutores são possíveis apenas devido aos efeitos quânticos. Mas mesmo algo tão modesto como uma ligação covalente - o componente básico de toda a química - é o resultado de interações quânticas de elétrons. Quando essas regras foram finalmente elaboradas na década de 1920, os cientistas perceberam que, pela primeira vez, foi criada uma teoria que detalhava o trabalho de toda a química. Em princípio, as equações quânticas poderiam simplesmente ser adaptadas para diferentes moléculas, resolvidas levando-se em consideração a energia do sistema e, então, determinando quais moléculas serão estáveis e quais reações ocorrerão espontaneamente. Mas,quando foi feita uma tentativa de sentar e calcular as soluções para essas equações, descobriu-se que isso é viável para o átomo mais simples (hidrogênio) e praticamente não para qualquer outro. Todos os outros cálculos se revelaram muito complicados.
O otimismo vertiginoso daqueles dias foi lindamente resumido por Paul Dirac:
Portanto, as leis físicas básicas necessárias para uma teoria matemática que descreveria a maior parte da física e toda a química já são conhecidas. O problema é que, na prática, a aplicação dessas leis fornece equações muito complexas, que somos objetivamente incapazes de resolver. Portanto, parece desejável desenvolver métodos aproximados para a aplicação prática da mecânica quântica.
1929
Muitos aceitaram o chamado de Dirac, e logo os físicos começaram a desenvolver métodos matemáticos que tornariam possível aproximar o comportamento das ligações moleculares e outros fenômenos químicos em um nível qualitativo. Tudo começou com uma descrição aproximada do comportamento dos elétrons - essa informação é estudada em um curso introdutório à química. Com essa descrição, cada elétron é trazido para seu próprio orbital, o que permite calcular a probabilidade de um elétron ser encontrado em um ponto específico nas proximidades de um núcleo atômico. Nesse caso, a forma de cada orbital depende da forma média de todos os outros orbitais. Visto que em tal descrição de acordo com o modelo de "campo autoconsistente", é assumido que cada elétron está ligado a apenas um orbital, esta imagem transmite de forma incompleta as propriedades reais dos elétrons. Ainda assim é o suficientepara determinar a energia total da molécula com um erro de apenas cerca de 0,5%.
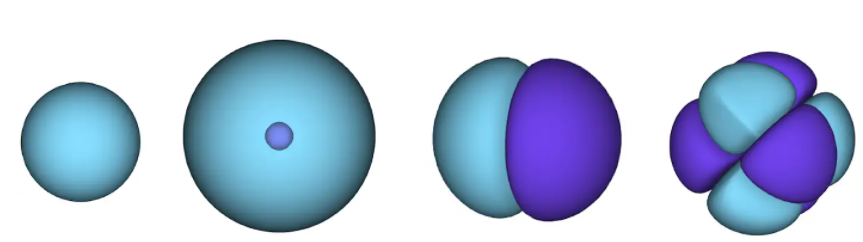
Figura 1 - orbitais atômicos. uma superfície é uma área na qual um elétron provavelmente está localizado. na região azul a função de onda é positiva e na violeta é negativa.
Infelizmente para o químico praticante, um erro de 0,5% é muito grande para ser tolerado. A energia das ligações moleculares é apenas uma pequena fração da energia total de um sistema, e a previsão correta de se uma molécula será estável pode muitas vezes depender de apenas 0,001% da energia total do sistema, ou cerca de 0,2% da energia de "correlação" restante.
Por exemplo, enquanto a energia total dos elétrons em uma molécula de butadieno é é quase 100.000 quilocalorias por mol, a diferença de energia entre as várias configurações possíveis da molécula é de apenas 1 quilocaloria por mol. Ou seja, se for necessário prever corretamente a forma natural da molécula de butadieno, o mesmo nível de precisão é necessário para medir a largura de um campo de futebol com uma precisão de um milímetro.
Com a proliferação da computação eletrônica logo após a Segunda Guerra Mundial, os cientistas desenvolveram uma série de métodos computacionais que não podiam ser descritos como campos autoconsistentes. Esses métodos são indicados por um conjunto inimaginável de abreviações que cobrem todo o alfabeto, mas cada um desses métodos contém algum tipo de compensação entre precisão e eficiência. Em um extremo estão os métodos que são, de fato, precisos, mas em escala pior do que exponencialmente conforme o número de elétrons aumenta - portanto, eles não são adequados para trabalhar com a maioria, mas com as menores moléculas. No outro extremo, existem métodos que escalam linearmente, mas não são muito precisos. Esses métodos computacionais tiveram um impacto tremendo na química prática - o Prêmio Nobel de Química de 1998 foi concedido aos autores de muitos desses algoritmos.
Apesar da amplitude das ferramentas de mecânica quântica computacional existentes, o problema de representar informações de forma eficiente exigiu o desenvolvimento de um novo método. Não é por acaso que apenas dezenas de milhares de elétrons estão envolvidos nos maiores cálculos químicos quânticos modernos (estamos falando dos métodos mais aproximados), enquanto os métodos clássicos de cálculos químicos, por exemplo, a dinâmica molecular, permitem lidar com milhões de átomos. Não é difícil descrever o estado de um sistema clássico - você só precisa rastrear a posição e o momento de cada partícula. Imaginar o estado de um sistema quântico é um desafio muito maior. Temos que atribuir um valor probabilístico a cada configuração possível das posições dos elétrons. Esta informação é codificada em uma função de onda,permitindo que você atribua um número positivo ou negativo para cada configuração de elétron, e a função de onda quadrada dá a probabilidade com a qual o sistema pode ser encontrado em tal configuração. O espaço de todas as configurações possíveis é colossal - se você tentasse imaginá-lo como uma grade com 100 pontos em cada dimensão, então o número de configurações possíveis de elétrons para um átomo de silício seria maior do que o número de átomos no universo!
É aqui que as redes neurais profundas são úteis. Nos últimos anos, enormes avanços foram feitos na representação de distribuições de probabilidade complexas com alta dimensionalidade usando redes neurais. Sabe-se agora como treinar efetivamente tais redes com a expectativa de sua escalabilidade. Sugerimos que como essas redes já provaram sua agilidade em treinar funções com várias dimensões na resolução de problemas da área de inteligência artificial, talvez funcionem para representar funções de ondas quânticas. Não fomos os primeiros a ter tais pensamentos - outros pesquisadores, em particular, Giuseppe Carleo e Matthias Troyerdemonstrou como o aprendizado profundo moderno é aplicável à solução de problemas quânticos idealizados. Queríamos usar redes neurais para resolver problemas mais realistas em química e física de estado sólido, o que significava que precisávamos levar os elétrons em consideração em nossos cálculos.
Há apenas uma advertência ao trabalhar com elétrons. Os elétrons devem obedecer ao princípio de exclusão de Pauli, ou seja, dois elétrons não podem estar simultaneamente no mesmo lugar. O fato é que os elétrons são partículas elementares dentre os férmions que constituem a maioria dos primeiros tijolos da matéria, em particular prótons, nêutrons, quarks, neutrinos, etc. A função de onda deles deve ser anti-simétrica - se você trocar dois elétrons, a função de onda é multiplicada por -1. Assim, há uma probabilidade zero de que dois elétrons fiquem um sobre o outro, uma vez que a probabilidade disso (e a função de onda correspondente) é igual a zero.
Portanto, foi necessário desenvolver uma rede neural de um novo tipo, que seria anti-simétrica em relação à entrada que entrava. Nós a batizamos de Rede Neural Fermiônica ou FermiNet. Na maioria dos métodos de química quântica, a antissimetria é introduzida usando uma função chamada determinante. O determinante é uma matriz que possui a seguinte propriedade: se você trocar duas de suas linhas, a saída é multiplicada por -1, exatamente como a função de onda dos férmions. Você pode pegar um monte de funções de um elétron, calculá-las para cada elétron em seu sistema e, em seguida, ajustar todos os resultados em uma matriz. Nesse caso, o determinante da matriz será uma função de onda verdadeiramente anti-simétrica. A principal limitação dessa abordagem é que a função resultante - chamada de Determinante Slater - não é amplamente aplicável.As funções de onda de sistemas reais são geralmente muito mais complexas. Normalmente, grandes combinações lineares de determinantes de Slater - às vezes milhões ou mais - são usadas para corrigir esse problema e, em seguida, algumas correções simples são feitas com base em pares de elétrons. Mesmo assim, o sistema pode não ser preciso o suficiente para calcular as energias.

2 – . – , 1. 1 2 , , -1. .
Redes neurais profundas são freqüentemente muito superiores em eficiência a combinações lineares de funções básicas ao representar funções complexas. No FermiNet, essa superioridade é alcançada pela introdução de cada uma das funções no determinante, a função de todos os elétrons. Este método é muito mais poderoso do que usar funções de um e dois elétrons. FermiNet fornece um fluxo de informações separado para cada elétron. Sem levar em conta quaisquer interações entre esses fluxos, a rede não seria mais expressiva do que o determinante usual de Slater. Para fazer mais, calculamos a média das informações coletadas de todos os fluxos em cada uma das camadas da rede e passamos essas informações para cada um dos fluxos para a próxima camada. Por conseguinte, tais fluxos têm propriedades de simetria adequadas para criar uma função anti-simétrica.
As informações sobre cada uma das camadas das redes neurais de grafos são agregadas de maneira semelhante . Ao contrário dos determinantes de Slater, as redes FermiNet são aproximadores de função universal , pelo menos enquanto as camadas das redes neurais permanecerem largas o suficiente. Isso significa que, se pudermos treinar essas redes corretamente, elas podem produzir uma solução quase exata para a equação de Schrödinger.
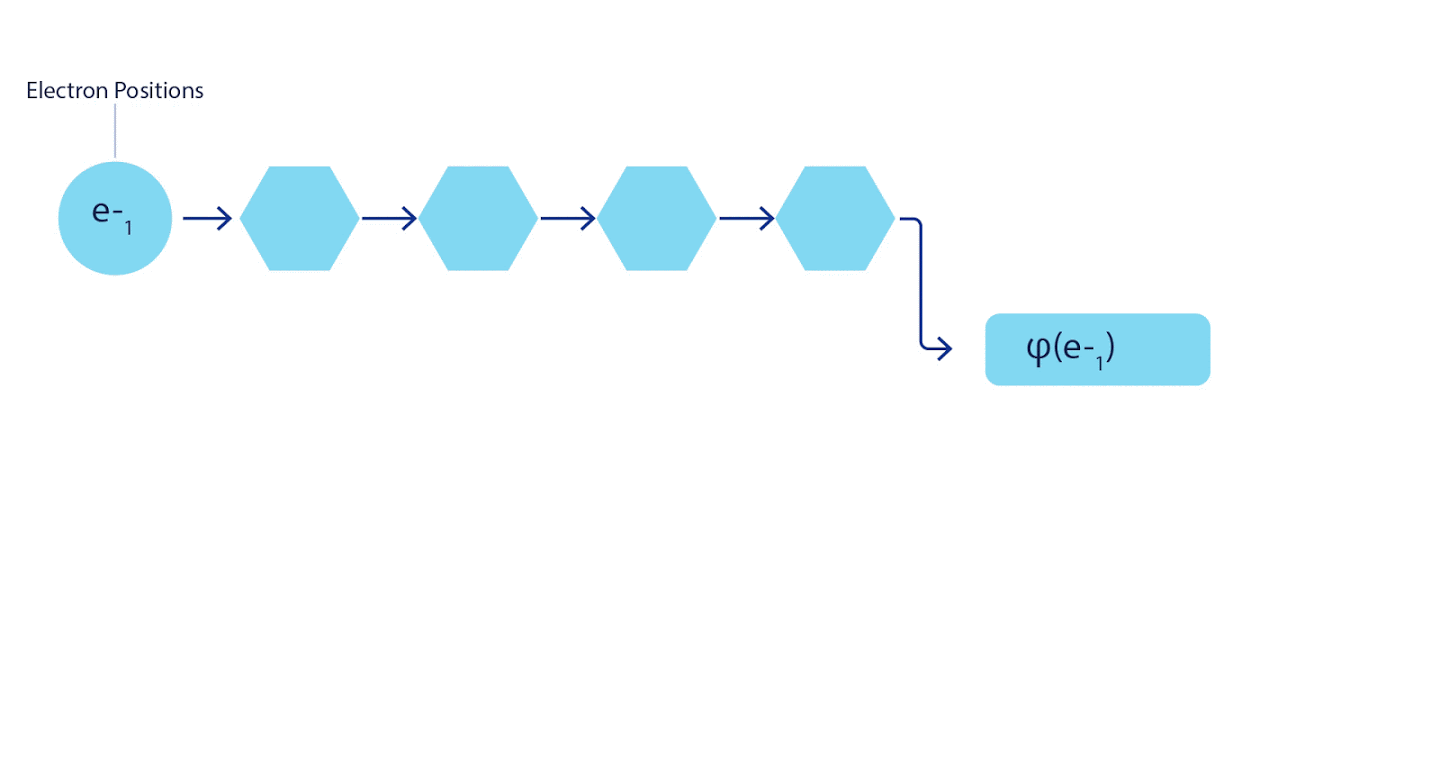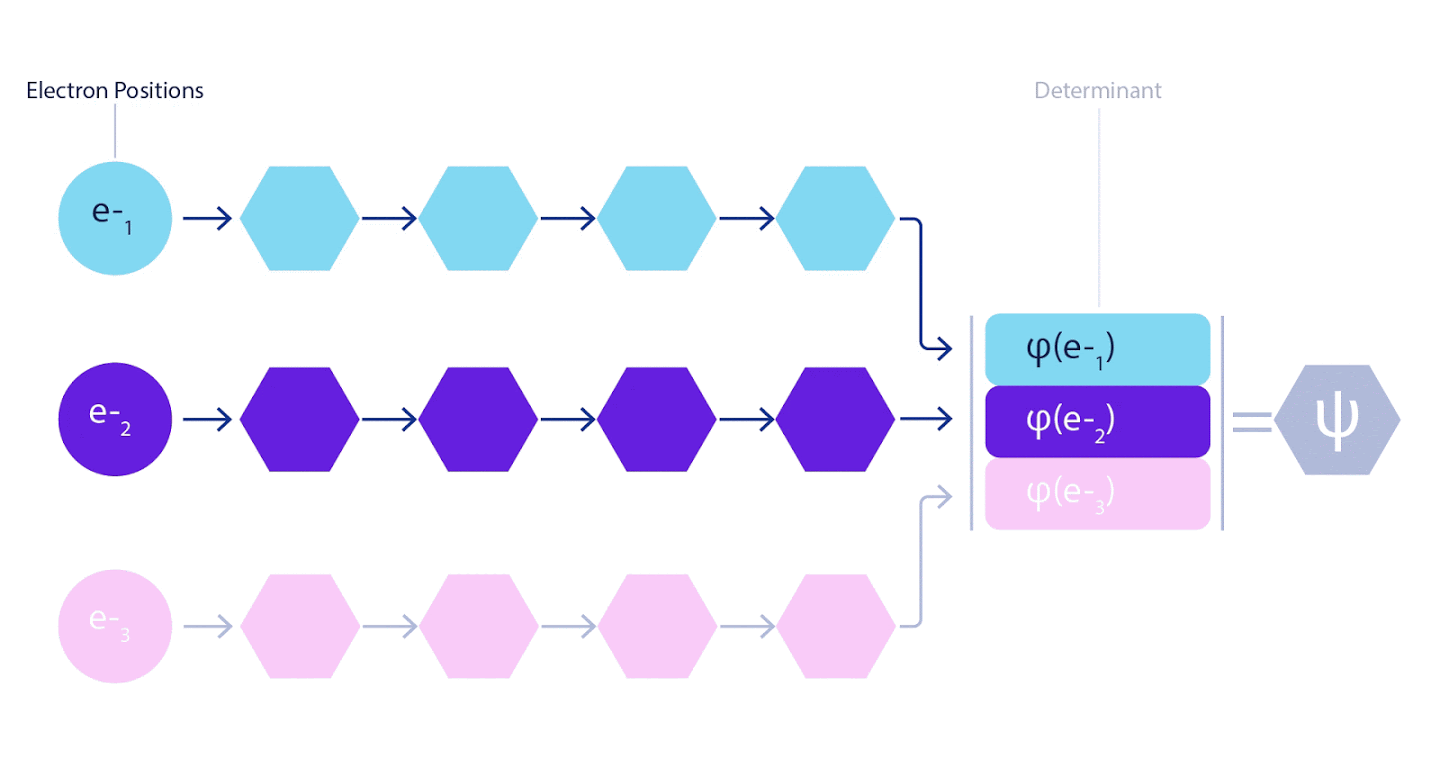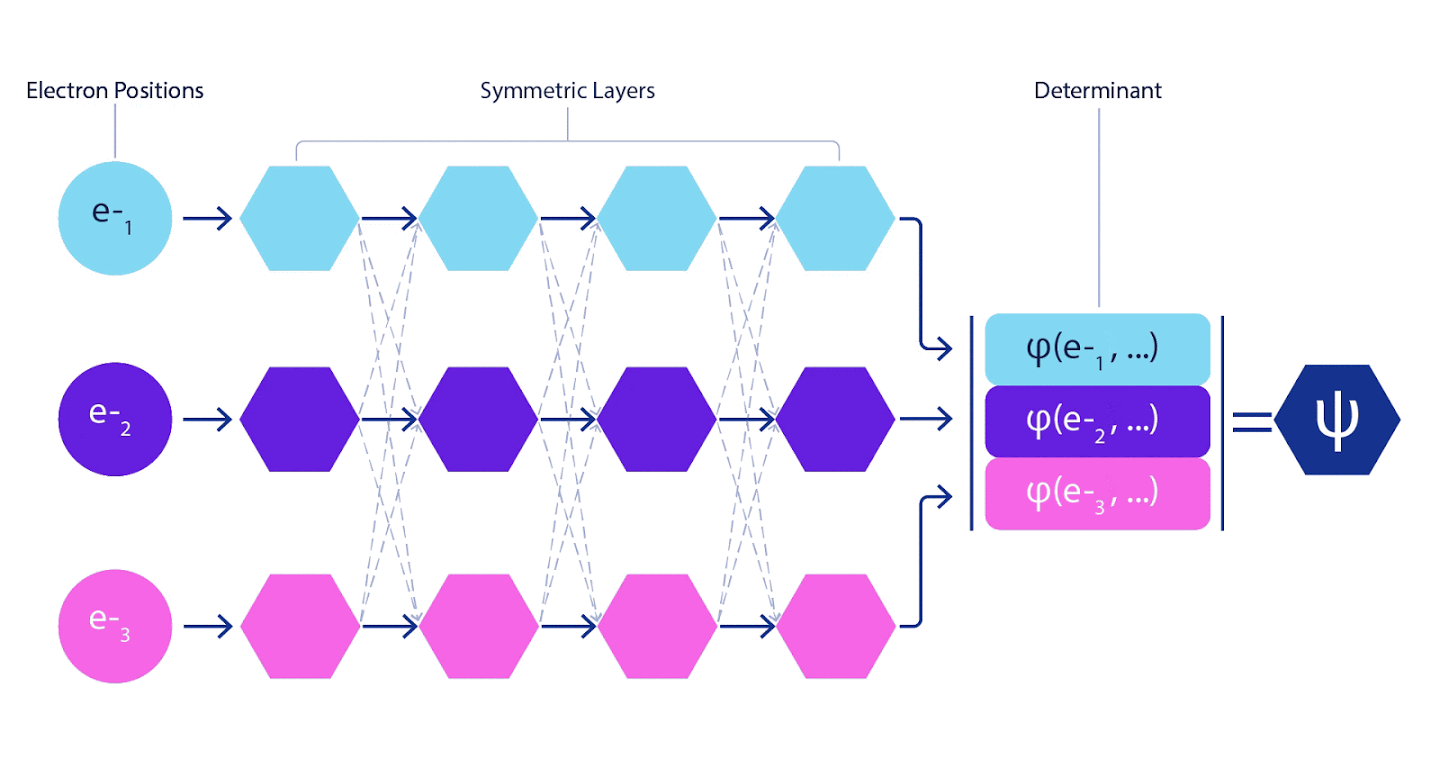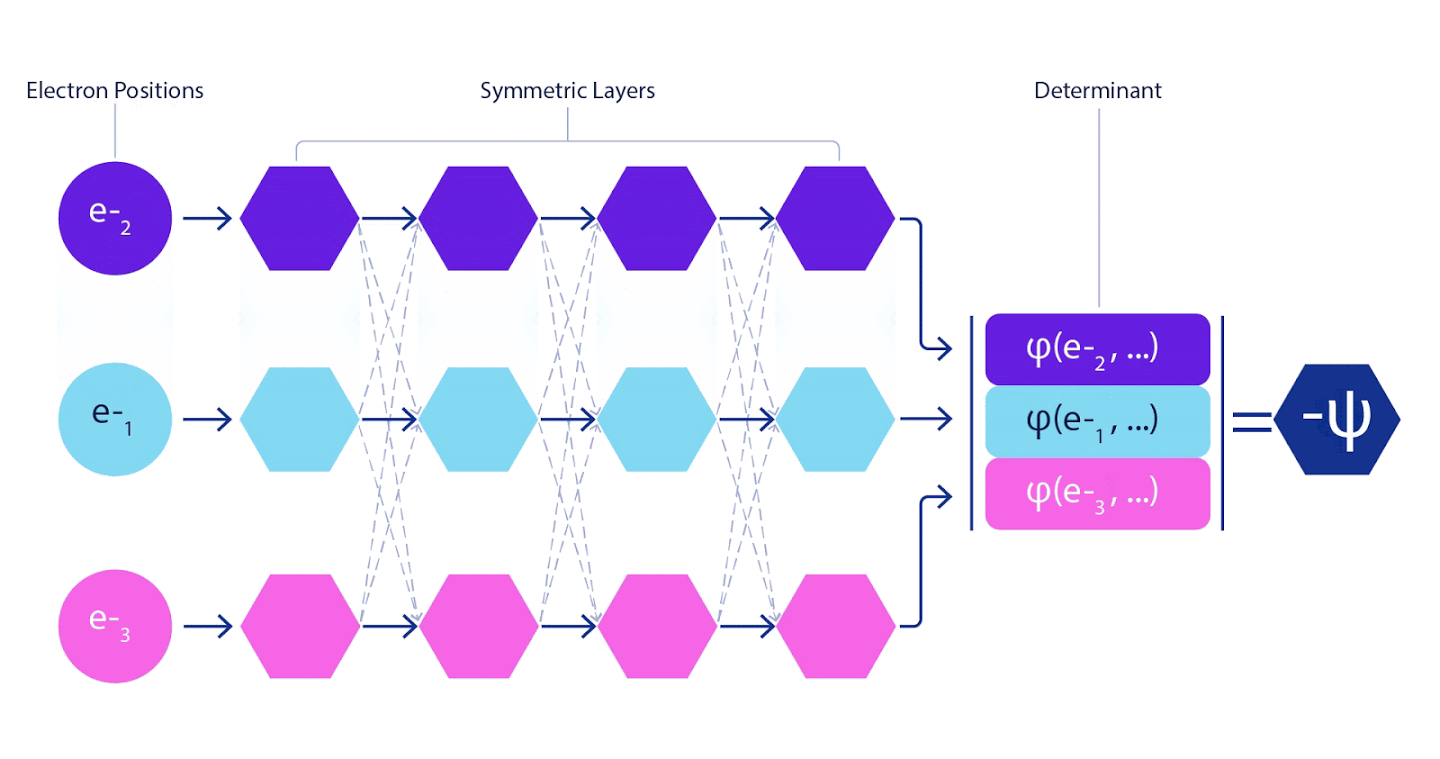
3 – FermiNet. (, ) . FermiNet , , , . , - -1.
Customizamos a rede FermiNet, minimizando a energia do sistema. Para fazer isso com precisão, precisaríamos calcular a função de onda em todas as configurações eletrônicas possíveis, portanto, teríamos que fazer isso aproximadamente. Portanto, pegamos uma amostra aleatória de configurações de elétrons, calculamos a energia localmente para cada variante de ordenação de elétrons e minimizamos essa energia, não a verdadeira. Esse método é chamado de "Monte Carlo" porque é um pouco como as ações de um jogador de cassino que joga os dados repetidamente. Como a função de onda quadrada permite observar alguma configuração de partículas em qualquer local, é mais conveniente gerar amostras da própria função de onda - em essência, simular o ato de observar partículas.
Embora a maioria das redes neurais seja treinada em alguns dados externos, em nosso caso a própria rede neural gera a entrada que vai para o treinamento. A situação é um pouco como se arrancar de um pântano pelos cabelos, e significa que não precisamos de nenhum dado de treinamento além das posições dos núcleos atômicos em torno dos quais os elétrons dançam. A ideia básica, conhecida como Método Variational Quantum Monte Carlo (ou VMC, para abreviar), existe na ciência desde 1960 e é geralmente considerada uma maneira barata, mas não muito precisa de calcular a energia de um sistema. Ao substituir funções de onda simples baseadas em determinantes de Slater por funções do FermiNet, conseguimos melhorar radicalmente a precisão dessa abordagem em todos os sistemas que consideramos.
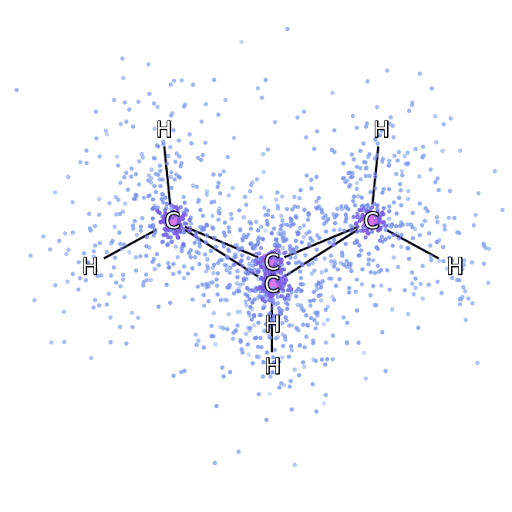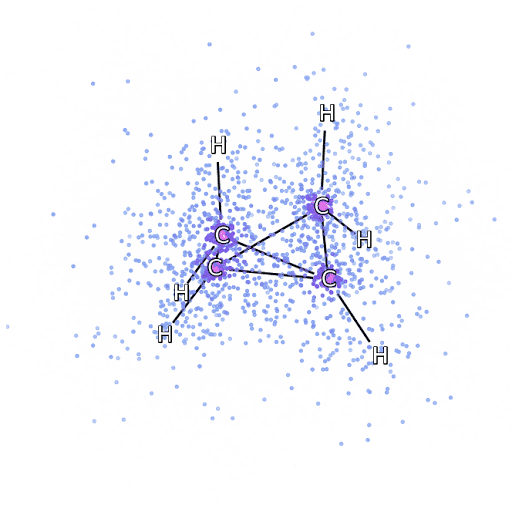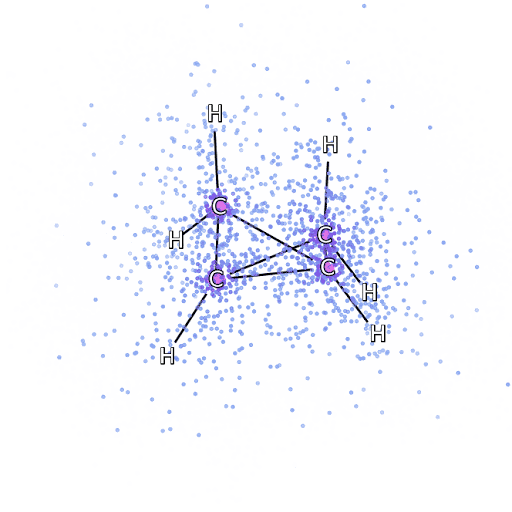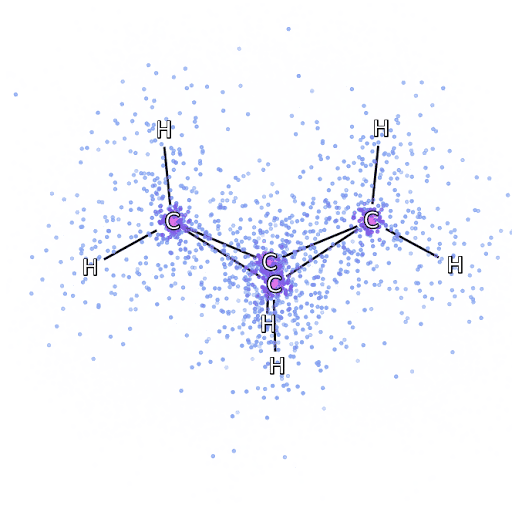
4 – , FermiNet, .
Para ter certeza de que o FermiNet é realmente uma inovação em sua área de assunto, começamos examinando sistemas simples e bem estudados, por exemplo, átomos da primeira linha da tabela periódica (de hidrogênio a neon). Esses são sistemas pequenos - 10 elétrons ou menos - então eles se prestam à pesquisa usando os métodos mais precisos (mas exponencialmente mais complexos). O FermiNet supera em muito os cálculos VMC comparáveis e muitas vezes pode reduzir o erro pela metade ou mais em comparação com cálculos exponencialmente escaláveis. Em sistemas maiores, os métodos que se tornam mais complexos exponencialmente tornam-se inaplicáveis, então usamos o método do cluster acoplado como referência. Este método funciona bem em moléculas com configurações estáveis, mas desliza quando as ligações são esticadas ou danificadas,e tais fatores são críticos para a compreensão das reações químicas. Embora tenha uma escala muito melhor do que exponencialmente, o método de agrupamento conectado usado neste estudo ainda funciona da melhor forma possível com moléculas de tamanho médio. Aplicamos FermiNet a moléculas cada vez maiores, de hidreto de lítio a biciclobutano - foi o maior sistema que vimos, com 30 elétrons. Nas menores moléculas, FermiNet capturou uma diferença surpreendente de 99,8% entre a energia dos aglomerados ligados e a energia derivada de um único determinante de Slater. No caso do biciclobutano, o FermiNet ainda capturou 97% ou mais dessa energia de correlação - uma grande conquista para uma abordagem supostamente "barata, mas imprecisa".Embora tenha uma escala muito melhor do que exponencialmente, o método de agrupamento conectado usado neste estudo ainda funciona da melhor forma possível com moléculas de tamanho médio. Aplicamos FermiNet a moléculas cada vez maiores, de hidreto de lítio a biciclobutano - foi o maior sistema que vimos, com 30 elétrons. Nas menores moléculas, FermiNet capturou uma diferença surpreendente de 99,8% entre a energia dos aglomerados ligados e a energia derivada de um único determinante de Slater. No caso do biciclobutano, o FermiNet ainda capturou 97% ou mais dessa energia de correlação - uma grande conquista para uma abordagem supostamente "barata, mas imprecisa".Embora tenha uma escala muito melhor do que exponencialmente, o método de agrupamento conectado usado neste estudo ainda funciona da melhor forma possível com moléculas de tamanho médio. Aplicamos FermiNet a moléculas cada vez maiores, de hidreto de lítio a biciclobutano - foi o maior sistema que vimos, com 30 elétrons. Nas menores moléculas, FermiNet capturou uma diferença surpreendente de 99,8% entre a energia dos aglomerados ligados e a energia derivada de um único determinante de Slater. No caso do biciclobutano, o FermiNet ainda capturou 97% ou mais dessa energia de correlação - uma grande conquista para uma abordagem supostamente "barata, mas imprecisa".que foi aplicado no estudo descrito, todos trabalham igualmente no máximo com moléculas de tamanho médio. Aplicamos FermiNet a moléculas cada vez maiores, de hidreto de lítio a biciclobutano - foi o maior sistema que vimos, com 30 elétrons. Nas menores moléculas, FermiNet capturou uma diferença surpreendente de 99,8% entre a energia dos aglomerados ligados e a energia derivada de um único determinante de Slater. No caso do biciclobutano, o FermiNet ainda capturou 97% ou mais dessa energia de correlação - uma grande conquista para uma abordagem supostamente "barata, mas imprecisa".que foi aplicado no estudo descrito, todos trabalham igualmente no máximo com moléculas de tamanho médio. Aplicamos FermiNet a moléculas cada vez maiores, de hidreto de lítio a biciclobutano - foi o maior sistema que vimos, com 30 elétrons. Nas menores moléculas, FermiNet capturou uma diferença surpreendente de 99,8% entre a energia dos aglomerados ligados e a energia derivada de um único determinante de Slater. No caso do biciclobutano, o FermiNet ainda capturou 97% ou mais dessa energia de correlação - uma grande conquista para uma abordagem supostamente "barata, mas imprecisa".do hidreto de lítio ao biciclobutano - foi o maior sistema que consideramos, tem 30 elétrons. Nas menores moléculas, FermiNet capturou uma diferença surpreendente de 99,8% entre a energia dos aglomerados ligados e a energia derivada de um único determinante de Slater. No caso do biciclobutano, o FermiNet ainda capturou 97% ou mais dessa energia de correlação - uma grande conquista para uma abordagem supostamente "barata, mas imprecisa".do hidreto de lítio ao biciclobutano - foi o maior sistema que consideramos, tem 30 elétrons. Nas menores moléculas, FermiNet capturou uma diferença surpreendente de 99,8% entre a energia dos aglomerados ligados e a energia derivada de um único determinante de Slater. No caso do biciclobutano, o FermiNet ainda capturou 97% ou mais dessa energia de correlação - uma grande conquista para uma abordagem supostamente "barata, mas imprecisa".mas imprecisa ".mas imprecisa ".

A Figura 5 é uma representação gráfica da fração da energia de correlação que FermiNet captura corretamente ao trabalhar com moléculas. A barra roxa marca 99% da energia de correlação. Da esquerda para a direita: hidreto de lítio, nitrogênio, etileno, ozônio, etanol e biciclobutano.
Embora os métodos de agrupamento acoplado funcionem bem com moléculas estáveis, a verdadeira "vanguarda" da química computacional tem a ver com a compreensão de como as moléculas se esticam, torcem e se quebram. Ao resolver esses problemas, os métodos de cluster conectado geralmente falham, então você deve comparar o resultado com o máximo de amostras de controle possível para ter certeza de que a resposta é consistente. No âmbito do experimento descrito, dois sistemas de controle alongado foram considerados - uma molécula de nitrogênio (N 2) e uma cadeia de hidrogênio de 10 átomos (H 10 ). Na molécula de nitrogênio, a ligação é especialmente complexa, pois nela participam 3 elétrons de cada átomo.
A cadeia de hidrogênio, por sua vez, é interessante para entender quais propriedades os elétrons apresentam nos materiais , por exemplo, para prever se um determinado material conduzirá eletricidade ou não. Em ambos os sistemas, o método de agrupamento conectado funcionou bem no equilíbrio, mas apresentou dificuldades quando as ligações foram esticadas. Os métodos VMC tradicionais não tiveram um bom desempenho em toda a gama de exemplos. Mas o FermiNet revelou-se um dos melhores métodos de todos investigados, independentemente do comprimento do link.
Conclusão
Acreditamos que o FermiNet é o início de grandes avanços na síntese de métodos de aprendizado profundo e química quântica computacional. A maioria dos sistemas com os quais FermiNet foi revisado até agora são bem compreendidos e compreendidos. Mas assim como os primeiros bons resultados usando aprendizado profundo em outros domínios estimularam uma onda de pesquisas adicionais e rápido progresso, esperançosamente o mesmo acontecerá com o FermiNet, e ideias para novas e ainda melhores arquiteturas de rede neural surgirão. Já depois que o trabalho descrito foi postado no arXiv, outros gruposcompartilharam suas abordagens para aplicar o aprendizado profundo para resolver problemas que envolvem muitos elétrons. Além disso, acabamos de nos aprofundar na física quântica computacional e planejamos usar o FermiNet para resolver problemas complexos no campo da ciência dos materiais e física do estado sólido.
O artigo científico está aqui , e o código pode ser visto aqui . Os autores gostariam de agradecer a Jim Kinwin, Adam Kine e Dominic Barlow por sua ajuda na preparação dos desenhos.