
Uma proteína da bactéria Staphylococcus aureus
No final de novembro, a equipe DeepMind do Google anunciou que seu sistema de aprendizado profundo AlphaFold alcançou níveis sem precedentes de precisão na resolução do problema de dobramento de proteínas , um problema difícil em bioquímica computacional.
Qual é o problema e por que é tão difícil de resolver?
As proteínas são longas cadeias de aminoácidos. Seu DNA codifica essas sequências, e o RNA ajuda a produzir proteínas de acordo com esse projeto genético. As proteínas são sintetizadas na forma de cadeias lineares, mas posteriormente dobram-se em estruturas esféricas complexas (veja a imagem no início do artigo).
Uma parte da corrente pode se enrolar em uma espiral fechada, " α-hélice . "A outra parte pode se dobrar para frente e para trás para formar uma figura larga e plana," folha β ":
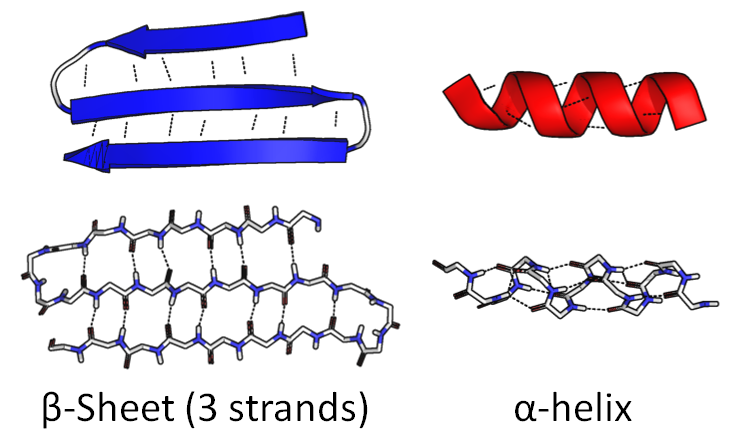
A própria sequência de aminoácidos é chamada de estrutura primária . Essas figuras são chamadas de estrutura secundária .
Esses próprios componentes também se dobram para formar formas complexas exclusivas. Isso é chamado de estrutura terciária :
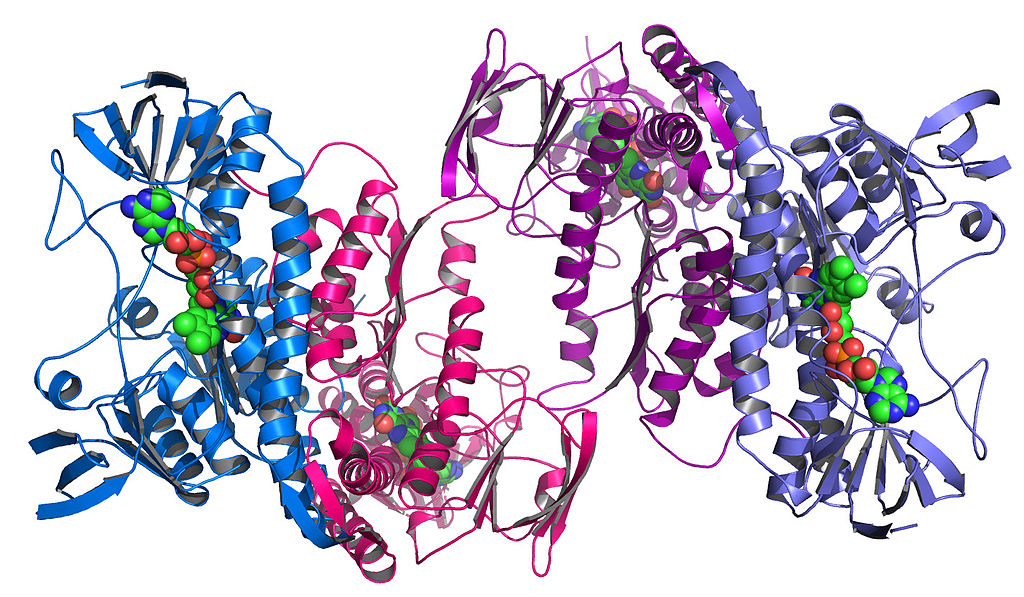
uma enzima retirada da

proteína Colwellia psicrerythraea RRM3
Parece bagunçada. Por que essa bola emaranhada de aminoácidos é tão importante?
A estrutura da proteína não é aleatória! Cada proteína se dobra em uma estrutura específica, única e amplamente previsível, que é essencial para seu funcionamento adequado. Devido à sua forma física, a proteína se adapta bem às estruturas com as quais pode se ligar. Outras propriedades físicas também são importantes, especialmente a distribuição de carga elétrica pela proteína. Na imagem, uma carga positiva é indicada em azul, uma carga negativa em vermelho:

Distribuição de carga superficial na proteína carreadora de lipídios das plantas 1 de arroz
Se uma proteína é, em essência, uma nanomáquina automontável, o objetivo principal de uma sequência de aminoácidos será produzir sua forma única, distribuição de carga e tudo o mais que determina a função da proteína. Como exatamente esse processo ocorre ainda não está totalmente claro - hoje é uma área ativa de pesquisa.
Em qualquer caso, entender a estrutura é importante para entender como ela funciona. No entanto, a sequência de DNA apenas define a estrutura primária da proteína. Como sabemos suas estruturas secundárias e terciárias - isto é, a forma exata que esse emaranhado terá?
Esse problema é chamado de problema de dobramento de proteínas e existem duas abordagens básicas para ele: medição e previsão.
Métodos experimentais podem medir a estrutura de uma proteína. No entanto, isso não é tão fácil de fazer: as estruturas não são visíveis ao microscópio óptico. Por muito tempo, a cristalografia de raios X foi o principal método de estudo de estruturas. Além disso, foi utilizada a ressonância magnética nuclear e, recentemente, surgiu uma nova tecnologia, a microscopia crioeletrônica .

Padrão de difração de raios-X da protease SARS
No entanto, esses métodos são caros, complexos e demorados e, além disso, não funcionam com todas as proteínas. Em particular, as proteínas incorporadas na membrana celular - o mesmo receptor da enzima 2 de conversão da angiotensina (ACE2) ao qual o vírus COVID-19 se liga - se dobra na bicamada lipídica células, e é muito difícil cristalizar.

A estrutura da membrana celular
Portanto, fomos capazes de desmontar a estrutura de uma pequena porcentagem das proteínas sequenciadas . O banco de dados universal de proteínas contém 180 milhões de sequências, enquanto o banco de dados de estruturas tridimensionais de proteínas contém apenas 170 mil posições.
Precisamos de um método melhor.
* * *
Lembre-se de que as estruturas secundária e terciária das proteínas são basicamente uma função da estrutura primária que conhecemos por meio do sequenciamento. E se, em vez de medir a estrutura de uma proteína, pudéssemos predizê-la?
Essa é a tarefa de prever a estrutura das proteínas. Os bioquímicos computacionais vêm trabalhando nisso há décadas.
Como você pode abordar isso?
A maneira óbvia é simular a física do processo diretamente. Simulamos forças para cada átomo, levando em consideração sua localização, carga e ligações químicas. Contamos as acelerações e velocidades e percorremos passo a passo a evolução do sistema. Isso é chamado de "dinâmica molecular".

Supercomputador " Anton " por DE Shaw Research

Supercomputador IBM Blue Gene
Online puzzle Foldit
O problema é que essa abordagem é extremamente intensiva em termos de computação. Uma proteína típica contém centenas de aminoácidos, ou seja, milhares de átomos. O ambiente também é importante: ao dobrar, a proteína interage com a água circundante. Portanto, é necessário simular o comportamento de cerca de 30 mil átomos. Nesse caso, ocorre uma interação eletrostática entre cada par de átomos, ou seja, com uma estimativa grosseira, obtemos 450 milhões de pares, um problema de complexidade O (N2). Existem algoritmos inteligentes que reduzem sua complexidade para O (N log N). Além disso, para simulação, é necessário calcular 10 9 -10 12 etapas. Dor de cabeça excepcional.
Ok, mas não precisamos simular todo o processo de dobramento. Outra abordagem sugere encontrar uma estrutura com energia potencial mínima. Os objetos geralmente tendem a ficar em repouso com o mínimo de energia, portanto, essa abordagem heurística é justificada. A energia pode ser calculada pelo mesmo modelo de dinâmica molecular, o que nos dá a magnitude das interações. Com essa abordagem, podemos testar vários candidatos e escolher a estrutura com o mínimo de energia. O problema, é claro, é de onde obter as estruturas. Eles são simplesmente muitos - o biólogo molecular Cyrus Levintol calculou que poderia haver cerca de 10.300 . Naturalmente, você pode usar uma abordagem mais inteligente do que a força bruta aleatória. Mas ainda existem muitos deles.
Portanto, muitas tentativas já foram feitas para acelerar esses cálculos. Anton, um supercomputador da DE Shaw Research, usa equipamentos especiais - circuitos integrados especiais. A IBM também está usando o supercomputador bio Blue Gene. Stanford lançou o projeto Folding @ Home, usando a potência distribuída dos computadores domésticos. O projeto Foldit da UW transformou o dobramento em um jogo para adicionar a intuição humana à computação.
Ainda assim, por muito tempo, nenhuma tecnologia foi capaz de prever uma ampla gama de estruturas de proteínas com alta precisão. Na competição CASP realizada duas vezes por ano, onde os resultados dos algoritmos são comparados com as estruturas medidas experimentalmente, os primeiros colocados receberam previsões com uma precisão de 30-40%. Até recentemente:

Melhor precisão preditiva mediana da equipe na categoria de modelagem livre.
Como o AlphaFold funciona? Ele usa várias redes neurais profundas para aprender diferentes funções associadas a cada proteína. Uma das funções principais é prever as distâncias resultantes entre pares de aminoácidos. Isso traz o algoritmo para a estrutura final. Em uma variante do algoritmo (descrito nos periódicos Nature and Proteins ), a função potencial dessa previsão foi derivada, à qual foi aplicada a descida gradiente mais simples, que funcionou surpreendentemente bem.
A principal vantagem do AlphaFold sobre os métodos anteriores é que ele não precisa fazer suposições sobre as estruturas. Alguns métodos funcionam dividindo as proteínas em seções, contando cada uma e depois juntando tudo de novo. AlphaFold não precisa disso.
Aparentemente, DeepMind considera o problema de dobramento resolvido, o que me parece uma simplificação exagerada, mas em qualquer caso, seu progresso é significativo. Especialistas não afiliados ao Google usam epítetos como “ fantástico ” e “ revolucionário ”.
A engenharia genética agora tem duas ferramentas poderosas, CRISPR e dobramento de proteínas. Talvez os anos 2020 sejam para a biotecnologia o que os anos 1970 foram para a computação.
Parabéns aos pesquisadores da DeepMind por esta descoberta!