Olá colegas! Não escrevo sobre Habr há cem anos, mas agora chegou a hora. Nesta primavera, ensinei o curso de ML avançado na MADE Big Data Academy do Mail.ru Group; Parece que os ouvintes gostaram, e agora me pediram para escrever não tanto um post publicitário, mas educacional sobre um dos tópicos do meu curso. A escolha foi quase óbvia: como exemplo de um modelo probabilístico complexo, discutimos um modelo SIR epidemiológico extremamente relevante (aparentemente ... mas mais sobre isso depois) que modela a propagação de doenças em uma população. Ele tem tudo: inferência aproximada por meio de métodos Markov Monte Carlo e modelos de Markov ocultos com o algoritmo estocástico de Viterbi e até dados apenas de presença.
Com este tópico, apenas uma pequena dificuldade surgiu: comecei a escrever sobre o que realmente contei e mostrei na palestra ... e de alguma forma rápida e imperceptivelmente vinte páginas de texto (bem, com imagens e código) acumuladas, o que ainda não é estava acabado e não era independente. E se você contar tudo de uma maneira que fique claro a partir do "zero" (não do zero absoluto, é claro), você poderá escrever cem páginas. Então, algum dia eu definitivamente irei escrevê-los, mas por agora apresento a sua atenção a primeira parte da descrição do modelo SIR, na qual podemos apenas colocar um problema e descrever o modelo do seu lado gerador - e se o público respeitado tiver interesse, então será possível Continuar.
Modelo SIR: declaração do problema
Eu amo meus amigos e eles me amam,
Nós somos tão próximos quanto podemos ser.
E só porque realmente nos importamos,
tudo o que recebemos, nós compartilhamos!
Tom Lehrer. Eu entendi de Agnes
Então, vamos descobrir. Informalmente, as premissas básicas do modelo SIR são as seguintes:
- existe uma certa população de pessoas na qual algum vírus terrível pode se espalhar;
- inicialmente, o vírus entra na população de uma forma ou de outra (por exemplo, aparece o primeiro doente), e então as pessoas se infectam umas com as outras;
- SIR , :
○ Susceptible ( , , .. ),
○ Infected ( )
○ Recovered ().
Para simplificar, vamos supor que aqueles que estiveram doentes sempre desenvolvem imunidade e são excluídos do ciclo do vírus na natureza. Conseqüentemente, as transições entre esses estados só podem ser da esquerda para a direita: Susceptível → Infectado → Recuperado.
A tarefa é, na verdade, treinar esse modelo, ou seja, entender algo sobre os parâmetros do processo de infecção a partir dos dados. É fácil entender como os dados se parecem - é simplesmente o número de infectados registrados em cada momento no tempo, que denotaremos pelo vetor... Por exemplo, quando dei meu dever de casa no curso sobre coronavírus (era, no entanto, sobre outros modelos), os dados para a Rússia eram assim:
[2,2,2,2,3,4,6,8,10,10,10,10,15,20,25,30,45,59,63,93,114,147,199,253,306,438,438,495,658,840,1036,1264,1534,1836,2337,2777,3548,4149,4731,5389,6343,7497,8672]Mas quais são os parâmetros deste modelo, como eles interagem entre si e como treiná-los, especialmente em um conjunto de dados tão pequeno, é uma questão mais séria.
Em geral, seguirei o esboço geral do trabalho ( Fintzi et al., 2016 ), no qual algoritmos MCMC são construídos para SIR, SEIR e algumas de suas variantes. Mas em comparação com ( Fintzi et al., 2016 ), simplificarei muito o modelo e sua apresentação. A principal simplificação é que ao invés do tempo contínuo, que é considerado na obra original, consideraremos o tempo discreto, ou seja, ao invés de um processo contínuo que em algum momento gera eventos da forma "infectado" e "recuperado", consideraremos que as pessoas passam por um conjunto de pontos discretos no tempo... Essa simplificação nos permitirá, em primeiro lugar, nos livrarmos de muitas dificuldades técnicas e, em segundo lugar, passar do algoritmo de Metrópolis-Hastings em geral para o algoritmo de amostragem de Gibbs, ou seja, não calcular a razão de metrópole, como é necessário fazer em ( Fintzi et al., 2016 ). Se você não entendeu o que acabei de dizer, não se preocupe: não precisaremos disso hoje, e se houver próximas partes, explicarei tudo lá.
Vamos denotar os estados do modelo SIR por S, I e R, respectivamente, e o estado de uma pessoa no momento - através ... Ao mesmo tempo, introduziremos imediatamente variáveis para o número total de pacientes que ainda não adoeceram.doente e recuperado no momento ... Homem total que teremosentão para qualquer um será executado ... E, como escrevi acima,deles são pessoas doentes registradas (testadas).
Os parâmetros desconhecidos do modelo sãoOnde:
- - a distribuição inicial dos casos; em outras palavras, para cada ; em nosso modelo simplificado, não vamos treinar, mas simplesmente registraremos 1-2 casos no primeiro momento;
- - a probabilidade de encontrar uma pessoa infectada na população em geral, ou seja, a probabilidade de uma pessoa no momento quando , serão detectados por meio de testes e inscritos nos dados ; em outras palavras, para cada pessoa doente, lançamos uma moeda para determinar se o teste vai encontrar ou não, então o item atual é tem uma distribuição binomial com parâmetros e :
- - a probabilidade de uma pessoa doente se recuperar em uma etapa de tempo, ou seja, a probabilidade de transição de um estado no estado ;
E - Este é o parâmetro mais interessante que mostra a probabilidade de infecção em uma contagem de tempo de uma pessoa infectada . Isso significa que, no modelo, assumimos que todas as pessoas da população "se comunicam" entre si ("comunicação" aqui significa contato suficiente para a transmissão da doença; o coronavírus é transmitido principalmente por gotículas aéreas, mas, é claro, é possível modelar e a propagação de qualquer outra doença; ver, por exemplo, a epígrafe), e a probabilidade de se infectar depende de quantos agora estão infectados, ou seja, do que está... Vamos assumir o modelo mais simples, em que a probabilidade de infecção de uma pessoa infectada é, e todos esses eventos são independentes, o que significa que a probabilidade de permanecer saudável é
Na verdade, essas suposições têm muito a discutir. O mais surpreendente aqui, na minha opinião pessoal, é a distribuição binomial para... Jogar uma moeda com probabilidade de detectar uma pessoa infectada é absolutamente normal, mas não é muito realista jogarmos essa moeda novamente a cada passo, ou seja, podemos “esquecer” uma pessoa infectada já detectada. Como resultado, o modelo SIR pode (e frequentemente o faz) produzir uma sequência completamente não monotônica; Isso é estranho, mas não parece ter um grande impacto no resultado, e seria muito mais difícil modelar esse processo de forma mais realista.
Além disso, é óbvio que este modelo se destina a uma população fechada, onde todos "se comunicam" com todos, então não faz sentido lançá-lo, digamos, para a Rússia com o parâmetro N de cem milhões ou para Moscou com o parâmetro dez milhões (e computacionalmente não funcionará). Uma área importante de extensões e extensões de modelos SIR é dedicada a isso: como adicionar um gráfico mais realista de interações e possíveis infecções; provavelmente falaremos sobre isso na última postagem.
Mas com todas essas simplificações, agora, usando os parâmetros acima, podemos escrever a matriz de probabilidade da transição entre os estados. Essas probabilidades (mais precisamente, especificamente a probabilidade de estar infectado) dependem das estatísticas gerais da população. Vamos designar o vetor de estados de todas as pessoas, exceto, através , e estender a mesma notação a todas as outras variáveis; por exemplo, É o número de infectados por vez , para não mencionar ... Então, para as probabilidades de transição de em Nós temos
e em todos os outros casos
O mesmo pode ser escrito mais claramente na forma de uma matriz de probabilidade de transição (a ordem das linhas e colunas é natural, SIR):
Para um leitor familiarizado com cadeias de Markov e modelos de Markov, pode parecer que este é apenas um modelo de Markov oculto: há um estado oculto, há uma distribuição clara de observáveis para cada estado oculto, as transições são realmente Markov, ou seja, o próximo estado oculto depende apenas do anterior ... Mas há , como se costuma dizer, há uma ressalva: as probabilidades de transição não podem ser consideradas constantes, elas mudam com a mudançae este é um aspecto extremamente essencial do modelo que não pode ser jogado fora.
Portanto, a inferência nesse modelo de repente se torna muito mais difícil do que no modelo oculto de Markov usual (embora nem sempre haja esse dom lá). No entanto, embora a conclusão seja mais complicada, ela ainda está sujeita à mente humana - nas próximas instalações (se houver) vou apenas falar sobre isso. E por hoje, quase o suficiente - agora vamos relaxar um pouco e tentar brincar com este modelo por enquanto em um sentido generativo.
Exemplo de geração de dados
Como grande conhecedor e especialista, um
introvertido entrou em quarentena.
***
“Eu não podia trabalhar em casa”, a
abelha tentou explicar para as minhocas.
***
Kaganov morreu brincando alegremente.
Claro, eu sinto muito por ele, entretanto ...
Leonid Kaganov. Quarentenas
Se os parâmetros do modelo são conhecidos e você só precisa gerar uma trajetória de como se desenvolveria a disseminação da doença na população, não há nada complicado no SIR. O código abaixo gera um exemplo de população de acordo com nosso modelo; afirma que codifiquei como, , ... Como avisei, para simplificar, irei supor que no momento tem exatamente um doente na população e depois continua.
def sample_population(N, T, true_rho, true_beta, true_mu):
true_log1mbeta = np.log(1 - true_beta)
cur_states = np.zeros(N)
cur_states[:1] = 1
cur_I, test_y, true_statistics = 1, [1], [[N-1, 1, 0]]
for t in range(T):
logprob_stay_healthy = cur_I * true_log1mbeta
for i in range(N):
if cur_states[i] == 0 and np.random.rand() < -np.expm1(logprob_stay_healthy):
cur_states[i] = 1
elif cur_states[i] == 1 and np.random.rand() < true_mu:
cur_states[i] = 2
cur_I = np.sum(cur_states == 1)
test_y.append( np.random.binomial( cur_I, true_rho ) )
true_statistics.append([np.sum(cur_states == 0), np.sum(cur_states == 1), np.sum(cur_states == 2)])
return test_y, np.array(true_statistics).T
N, T, true_rho, true_beta, true_mu = 100, 20, 0.1, 0.05, 0.1
test_y, true_statistics = sample_population(N, T, true_rho, true_beta, true_mu)
Este código fornece não apenas os dados reais mas também estatísticas "verdadeiras" , , para que possam ser visualizados. Vamos fazer isso; para parâmetros, , , , você pode obter algo assim:
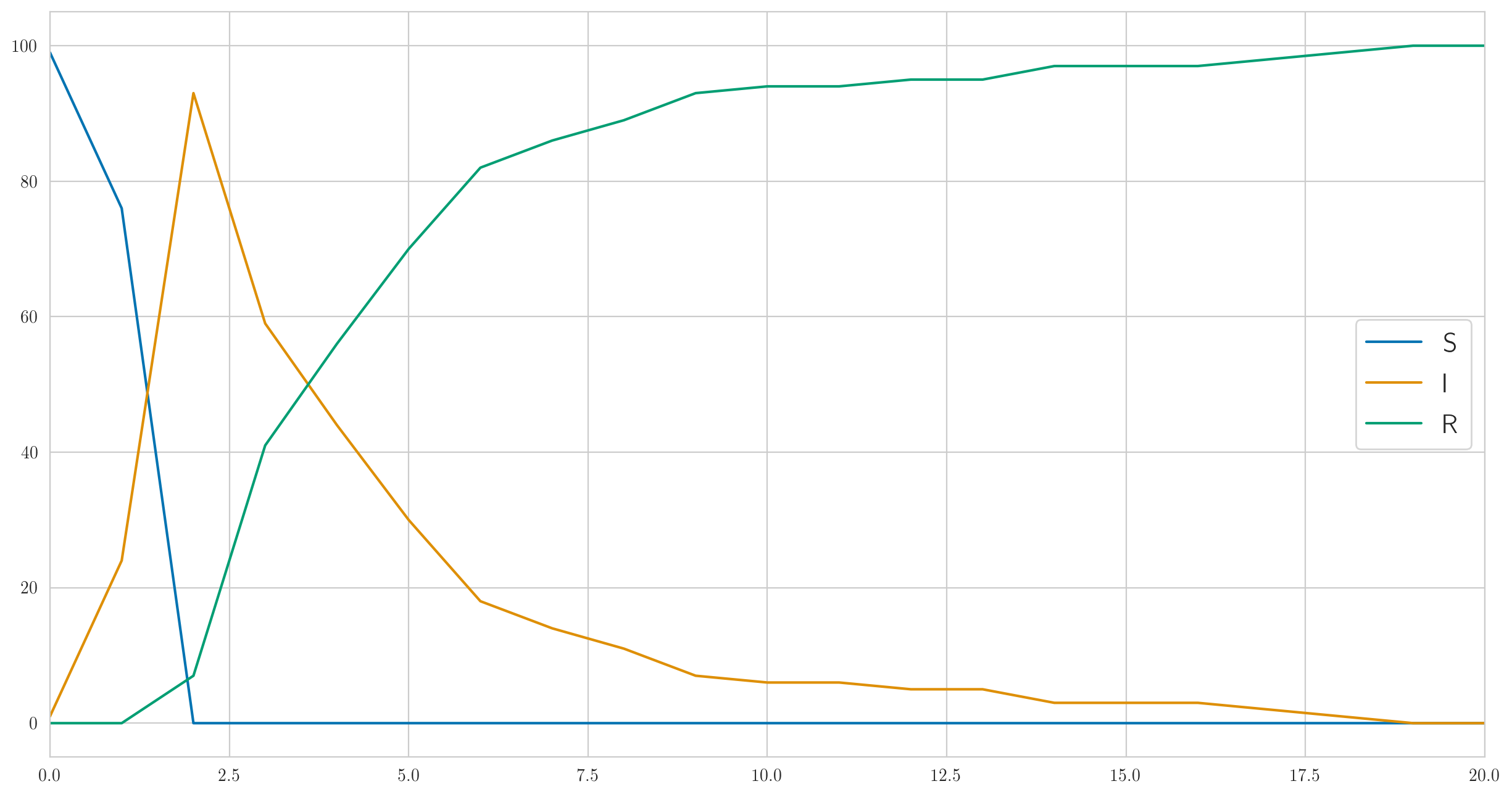
Como você pode ver, neste exemplo, todo mundo fica doente muito rapidamente e, lentamente, começa a melhorar. E os dados reais observados sobre os doentes são assim:
[1 6 13 6 6 4 1 0 0 1 0 1 2 0 0 0 0 0 0 1 0 0]
Observe que, como discutimos acima, não há monotonia aqui.
Mas esse certamente não é o único comportamento possível. Escolhi os parâmetros acima para que a infecção fosse rápida o suficiente e a doença abrangesse quase imediatamente todas as cem pessoas em nossa população de teste. E se você fizer menor, por exemplo , então a infecção será muito mais lenta e nem todos terão tempo de adoecer antes que o último doente se recupere e não fique. Algo assim:
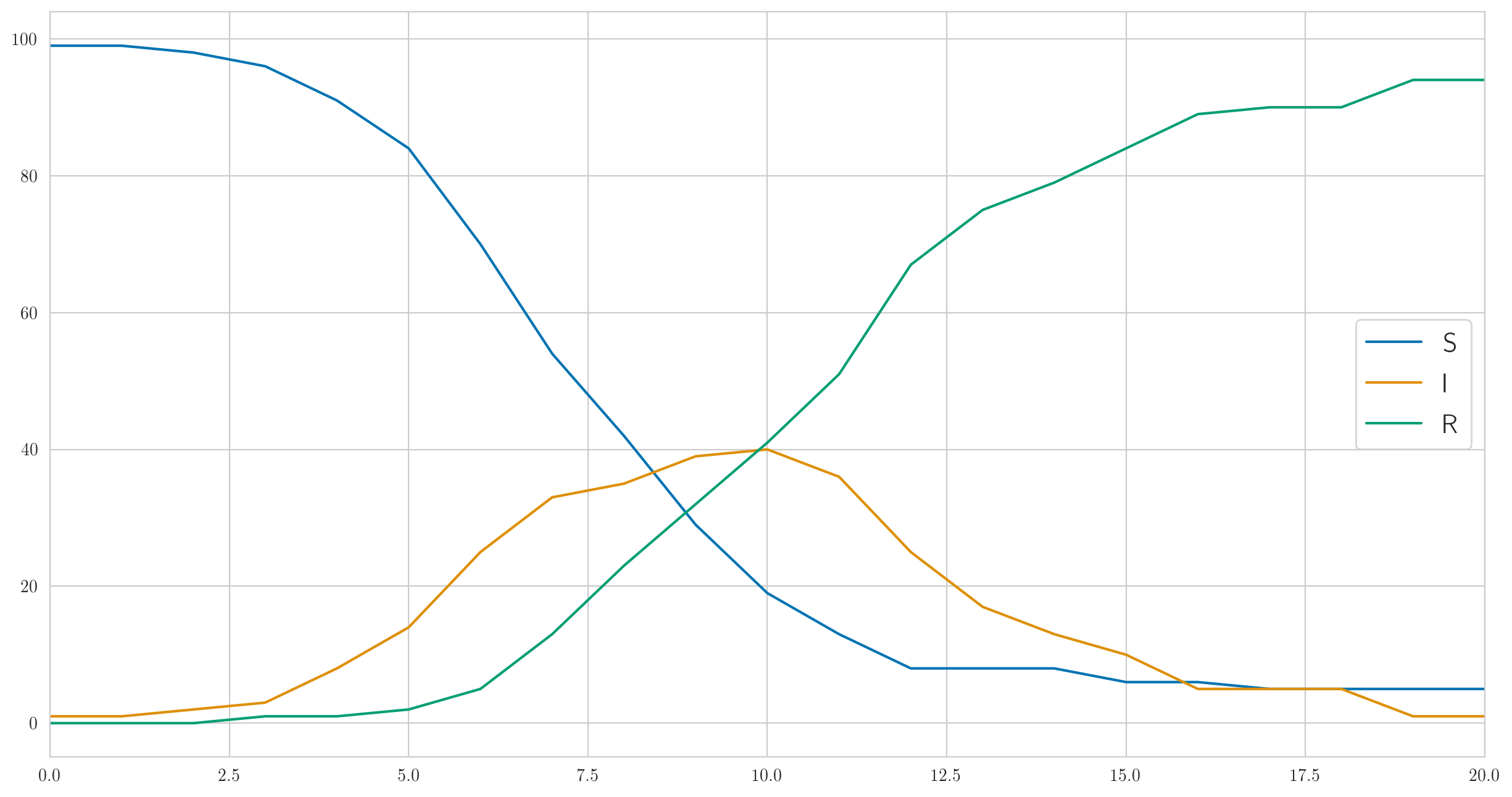
E o número de casos detectados também é completamente diferente:
[1 0 0 0 0 1 2 2 3 1 1 9 3 1 3 1 0 1 0 0 0]
É possível "estrangular" ainda mais a doença; por exemplo, vamos sairmas coloque , ou seja, a cada intervalo de tempo, cada doente tem uma chance de se recuperar em 0,5 (no final, isso é lógico: ele vai se recuperar ou não). Então, apenas 50-60 em 100 pessoas ficarão doentes com o terrível vírus; curvas podem ser assim:

[1 0 0 1 3 2 1 1 0 3 0 0 0 0 0 1 0 0 0 0 0]
Mas o quadro geral, é claro, em qualquer caso, parece o mesmo: primeiro, crescimento exponencial e depois saturação; a única diferença é se a última operadora tem tempo para se recuperar antes que todos sejam reinicializados.
Bem, é hora de resumir os resultados provisórios. Hoje vimos como é um dos modelos epidemiológicos mais simples. Apesar de muitas suposições simplistas, o modelo SIR ainda é muito útil mesmo nessa forma; o fato é que a maioria de suas extensões são bastante diretas e não mudam a essência geral da questão. Portanto, se continuarmos, na próxima série irei falar sobre como treinar um modelo SIR; no entanto, isso envolverá tantas coisas interessantes que provavelmente não caberá em uma postagem também. Até a próxima vez!